Approval Date:
Oxitinib Mesylate TabletsInstructions
Please read the instructions carefully and use under the guidance of your physician
[Medication Name]
Generic name: Ocitinib Mesylate Tablets
Trade name: TARISSA®/TAGRISSO®
English name: Osimertinib Mesylate Tablets
Hanyu Pinyin: Jiahuangsuan Aoxitini Pian
[Ingredients].
The active ingredient of this product is Osimertinib Mesylate
Chemical name: N-{2-{[2-(dimethylamino)ethyl](methyl)amino}-4-methoxy-5-{[4-(l-methyl-lH-indol-3-yl)pyrimidin-2-yl]amino}phenyl)propan-2-enamide methanesulfonate
Chemical structure formula: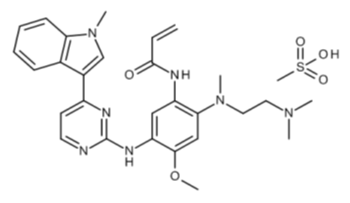
Molecular formula: C28H33N7O2 . CH4O3S
Molecular weight: 595.71
[Properties].
This product is a light brown film-coated tablet, which appears white to light brown after removing the coating.
Ositinib mesylate 40 mg: one side is printed with “AZ” and “40”, the other side is blank.
Ocitinib Mesylate 80 mg: One side is printed with “AZ” and “80”, the other side is blank.
[Indications
[Indications].
This product is indicated for the treatment of adult patients with locally advanced or metastatic non-small cell lung cancer (NSCLC) who have experienced disease progression on or after prior epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor (TKI) therapy and have been tested positive for EGFR T790M mutations.
[Specifications]
[Dosage and Administration]
This product should be prescribed by a physician experienced in antineoplastic therapy.
The status of the EGFR T790M mutation first needs to be clarified before using this product for the treatment of locally advanced or metastatic NSCLC. The presence of an EGFR T790M mutation should be determined using a well-validated assay before treatment with this product (see [Precautions] for details).
Dosage
The recommended dose of this product is 80 mg daily until disease progression or intolerable toxicity occurs.
If 1 dose of this product is missed, the product should be made up unless the next dose is within 12 hours.
This product should be taken at the same time each day, either with a meal or on an empty stomach.
Dose Adjustment
Dosing may be suspended or reduced depending on the safety and tolerability of the individual patient. If a dose reduction is required, the dose should be reduced to 40 mg once daily.
See Table 1 for principles of dose reduction after the occurrence of adverse events (AEs) and toxicity.
Table 1. principles of dose adjustment of oseltinib mesylate tablets after the occurrence of an adverse event
| AdverseEventa | Dose adjustment | |
| Lungs | Interstitial lung disease/non-infectious pneumonia | Permanent discontinuation of this product |
| Heart | At least two separate ECG tests suggesting a QTc interval greater than 500 ms | Suspend until QTc interval is less than 481 ms or return to baseline level (if baseline value is greater than or equal to 481 ms) restart dosing with 40 mg |
| Prolonged QTc interval with signs or symptoms of severe arrhythmia | Permanent discontinuation of this product | |
| Asymptomatic absolute decrease in left ventricular ejection fraction (LVEF) of 10% and less than 50% relative to baseline | Suspension of therapy with this product for up to 4 weeks. – If improvement is to baseline LVEF levels, restart treatment. – If no improvement to baseline levels, permanently discontinue treatment. |
|
| Symptomatic Congestive Heart Failure | Permanent discontinuation of therapy with this product. | |
| Other | Grade 3 or higher adverse reactions | Discontinue use of this product for up to 3 weeks |
| then the drug may be resumed at the original dose (80 mg) or at a reduced dose (40 mg). | ||
| If grade 3 or higher adverse reactions do not decrease to grade 0-2 after 3 weeks of suspension of this product | Permanent discontinuation of this product |
a Note: The intensity of clinical adverse events is graded according to the National Cancer Institute (NCI) Common Terminology Criteria for Adverse Events (CTCAE), version 4.0.
QTc: QT interval corrected by heart rate
Special Populations
No dose adjustment for patient age, weight, gender, race, or smoking status is required (see [Pharmacokinetics]).
Hepatic Impairment
No dose adjustment is required in patients with mild hepatic impairment (total bilirubin< upper limit of normal (ULN) and glutamic oxalacetic transaminase (AST) of 1 to 1.5 x ULN; or total bilirubin of 1 to 1.5 x ULN with unlimited AST), but this product should still be used with caution in such patients. The safety and efficacy of this product in patients with moderate to severe hepatic impairment are not known. Until more information is available, this product is not recommended for use in patients with moderate to severe hepatic impairment. (See [Pharmacokinetics]).
Renal Impairment
No dose adjustment is required for the use of this product in patients with mild to moderate renal impairment. There are limited data on the use of this product in patients with severe renal impairment. The safety and efficacy of this product in patients with end-stage renal disease (creatinine clearance (CLcr) <15 mL/min as calculated by the Cockcroft and Gault equation) or who are on dialysis is unknown. This product should be used with caution in patients with severe or end-stage renal impairment (see [Pharmacokinetics]).
Method of Administration
This product is for oral use. This product should be taken whole and with water and should not be crushed, broken, or chewed.
If the patient is unable to swallow the medication, the tablet may be dissolved in 50 mL of carbonate-free water. The tablets should be put into the water without crushing, stirred directly until dispersed and swallowed quickly. An additional half cup of water should be added to ensure that no residue remains in the cup, followed by rapid consumption. No other liquids should be added.
When feeding via the gastric tube is required, it can be handled in the same manner as above, except that 15 mL of water is used for the initial dissolution of the drug and 15 mL for the subsequent rinsing of the residue. all 30 mL of this liquid should be fed according to the instructions of the manufacturer of the nasogastric tube and rinsed with an appropriate amount of water. Both these dissolution solutions and the residual solution should be administered within 30 minutes of adding the tablets to the water.
[Adverse Reactions]
Summary of Safety Data(Not Considering Cause and Effect)
Safety data were obtained in two global single-arm clinical trials (Phase II portion of the AURA extension study and the AURA 2 study) in 411 previously treated patients with T790M mutation-positive NSCLC taking a dose of 80 mg daily. 333 of the 411 patients were exposed to treatment with this product for at least 6 months; 97 patients were exposed for at least 9 Of the 411 patients, 333 were exposed for at least 6 months; 97 were exposed for at least 9 months; however, no patients were exposed for up to 12 months.
The most common (>20%) adverse events in patients in the treatment group were diarrhea (42%), rash (41%), dry skin (31%), and finger (toe) nail toxicity (25%).
The most common adverse events leading to dose reduction or treatment interruption were prolonged ECG QTc interval (2.2%) and neutropenia (1.9%). serious adverse events reported in 2% or more patients were pneumonia and pulmonary embolism. Other fatal adverse events reported in more than 1 patient included infectious pneumonia (4 patients) and cardiovascular accident/cerebral hemorrhage (2 patients). Treatment was discontinued due to adverse events in 5.6% of patients in the treatment group. The most common adverse events leading to discontinuation of treatment were interstitial lung disease/non-infectious pneumonia and cerebrovascular accident/cerebral infarction.
Table2 Occurrence>10% of allNCICTCAE* in two global single-arm studiesGrade Adverse Events and Incidence>2% ofNCI CTCAE* Grade 3-4 Adverse Events
| AdverseEvent | Oxitinib N=411 |
|
| All levels | 3~4Levels f | |
| % | % | |
| Gastrointestinal Disorders | ||
| 42 | 1.0 | |
| Nasty | 17 | 0.5 |
| Nag | 16 | 0.7 |
| 15 | 0.2 | |
| Stomatitis | 12 | 0 |
| Dermatosis | ||
| 41 | 0.5 | |
| Dry skin b | 31 | 0 |
| Finger (toe) nail toxicity c | 25 | 0 |
| itching | 14 | 0 |
| Eye disease d | 18 | 0.2 |
| Respiratory disease | ||
| 14 | 0.2 | |
| Systemic disease | ||
| 14 | 0.5 | |
| 13 | 0.7 | |
| Central nervous system disorders | ||
| 10 | 0.2 | |
| Infection | ||
| 4 | 2.2 | |
| Vascular events | ||
| 7 | 2.4 | |
* NCI CTCAE v4.0.
- Reported cases that include the following rash-like categorization terms: rash, generalized rash, erythematous rash, macular rash, maculopapular rash, papules, pustular rash, erythema, folliculitis, acne, dermatitis, and acneiform dermatitis.
- Including dry skin, eczema, cracked skin, and dry disease.
- Includes reported cases of the following categorized terms: nail bed disease, nail bed inflammation, nail bed tenderness, nail bed discoloration, finger (toe) nail disease, finger (toe) nail toxicity nail, finger (toe) nail atrophy, finger (toe) nail infection, finger (toe) nail sclerosis, brittle nail, nail detachment, nail loss, and nail fungus.
- Including dry eyes, blurred vision, keratitis, cataracts, eye irritation, blepharitis, eye pain, increased tearing, and mosquito flying. Patients presenting with other ocular toxicity<1%.
- Including deep vein thrombosis, internal jugular vein thrombosis, and pulmonary embolism.
- No grade 4 events were reported.
.
Summary of Safety Data(Parts Specified as Adverse Drug Reactions)
Table 3 lists the incidence of common adverse drug reactions (ADRs) in patients taking this product.
Adverse reactions are tabulated according to MedDRA’s Systemic Organ Classification (SOC). Within each SOC, ADRs were ranked by frequency of occurrence, with the most frequent ADRs leading the list. Within each frequency category, ADRs were ranked in descending order of severity. In addition, the corresponding frequency of each ADR was categorized according to the conventional concept of CIOMS III, and these frequency categories were: very common (≥1/10); common (>1/100 to <1/10); rare (≥1/1,000 to <1/100); rare (≥1/10,000 to <1/1,000) ; extremely rare (<1/10,000); unknown (cannot be estimated based on available data). This section only incorporates data obtained from concluded studies in which patient exposure was known.
Table 3. adverse drug reactions reported during the AURAa study
| MedDRA Terms | CIOMSClassification/Overall Frequency (AllCTCAE classification)b | Frequency of Grades 3-4CTCAE | |
| Respiratory, thoracic and mediastinal system disorders | Interstitial lung diseasec | Common (2.7%)d |
0.7% |
| Gastrointestinal disorders | Diarrhea | Very common (42%) | 1% |
| Stomatitis | Very common (12%) | 0% | |
| Dermal and subcutaneous tissue disorders |
Rashe | Very common (41%) |
0.5% |
| Dry skinf | Very common (31%) |
0% | |
| Glenoid nail g | Very common (25%) | 0% | |
| itchingh | Very common (14%) | 0% | |
| Laboratory tests(Determined based on test results, and by change in CTCAE level is given) | Prolonged QT interval i | Rare (0.2%) | |
| Decreased platelet countj | Very common (54%) | 1.2% | |
| Leukopeniaj | Very common (67%) | 1.2% |
|
| Neutropeniaj | Very common (33%) | 3.4% | |
a The data presented in the table are cumulative data obtained from the AURA Extension Study (AURA ex; Phase II) and the AURA 2 Study; only adverse events occurring in patients who took at least 1 dose of this product are summarized.
b National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE) version 4.0.
c Reported cases that include the following categorical terms: interstitial pneumonia and noninfectious pneumonia.
d There were 4 reported cases of CTCAE grade 5 events (fatal events).
e Included are reported cases with the following categorization terms for rash-like events: rash, panniculitis, erythema, maculopapular rash, papule, pustular rash, erythema, folliculitis, acne, dermatitis, and acneiform dermatitis.
f Includes reported cases of the following categorized terms: dry skin, chapped skin, dry disease, eczema.
g Reported cases that include the following categorical terms: nail bed disease, nail bed inflammation, nail bed tenderness, nail bed discoloration, finger (toe) nail disease, finger (toe) nail toxicity, finger (toe) nail atrophy, finger (toe) nail infection, finger (toe) nail sclerosis, brittle nail, nail detachment, demodicosis, and nail fungus.
h Includes reported cases of the following categorized terms: pruritus, generalized pruritus, eyelid pruritus.
i Indicates patients with prolonged QTcF >500msec (calculated from ECG data, not the incidence of reported adverse events).
j indicates the incidence seen on laboratory tests, not the incidence of reported adverse events.
AURA 17 Summary of Safety Data
Safety data for the Asia-Pacific population of 171 (148 of whom were Chinese patients) previously treated T790M mutation-positive NSCLC patients taking a dose of 80 mg daily were obtained in the Asia-Pacific phase II study (Table 4. AURA 17, see [Clinical Trials]). safety data from AURA 17 were consistent with global phase II safety The safety data from AURA 17 were consistent with the global phase II safety data. The majority of adverse reactions were Grade 1 or 2 in severity. The most commonly reported ADRs were diarrhea (29%) and rash (20%), and the incidence of CTCAE grade 3 or higher adverse events in the AURA 17 study was 14%. Among patients receiving this product on a daily regimen of 80 mg, 0.6% were reduced due to ADRs. There were 1.2% of patients who discontinued the drug early due to adverse reactions or abnormal laboratory tests.
Table4. AURA 17 aAdverse Drug Reactions Reported During the Study
| MedDRA Terms | CIOMSClassification/Overall Frequency (AllCTCAE classification)b | Frequency of Grades 3-4CTCAE | |
| Respiratory, thoracic and mediastinal system disorders | Interstitial lung diseasec | Common (1.8%)d |
0% |
| Gastrointestinal Disorders | Diarrhea | Very common (29%) | 0% |
| Stomatitis | Common (3.5%) | 0% | |
| Dermal and subcutaneous tissue disorders |
Rashe | Very common (20%) |
0% |
| Dry skinf | Very common (17%) |
0.6% | |
| Glenoid nail g | Common (7.6%) | 0% | |
| itchingh | Very common (13%) | 0% | |
| Laboratory tests(Determined based on test results, and by change in CTCAE level is given) | QTc interval prolongationi | Very rare (0%) | |
| Very common (65%) | 1.2% | ||
| Leukopeniaj | Very common (67%) | 0% |
|
| Neutropeniaj | Very common (29%) | 1.2% | |
a The data presented in the table are based on the first data cutoff date of the AURA 17 study. At this point in time, all patients had the opportunity to receive 18 weeks (4.5 months) of treatment; only adverse events that occurred in patients who took at least 1 dose of this product are summarized.
b National Cancer Institute Common Terminology Criteria for Adverse Events, version 4.0.
c Reported cases that include the following categorical terms: interstitial pneumonia and noninfectious pneumonia.
d There was one reported case of CTCAE grade 5 event (fatal event).
e Included are reported cases with the following categorization terms for rash-like events: rash, panniculitis, erythema, maculopapular rash, papule, pustular rash, erythema, folliculitis, acne, dermatitis, and acneiform dermatitis.
f Includes reported cases of the following categorized terms: dry skin, chapped skin, dry disease, eczema.
g Reported cases that include the following categorical terms: nail bed disease, nail bed inflammation, nail bed tenderness, nail bed discoloration, finger (toe) nail disease, finger (toe) nail toxicity, finger (toe) nail atrophy, finger (toe) nail infection, finger (toe) nail sclerosis, brittle nail, nail detachment, demodicosis, and nail fungus.
h Includes reported cases of the following categorized terms: pruritus, generalized pruritus, eyelid pruritus.
i indicates patients with QTcF >500msec (calculated from ECG data, not the incidence of reported adverse events)
j indicates the incidence seen on laboratory tests, not the incidence of reported adverse events.
Description of Specific Adverse Drug Reactions
Interstitial Lung Disease(ILD)
ILD occurred in 6.2% of patients of Japanese descent during the phase II study, compared with 1.2% of non-Japanese Asian patients and 2.4% of non-Asian patients. the median time to onset of ILD or ILD-like adverse reactions was 2.7 months (see [Precautions]).
QTcProlonged interval
Of the 411 patients in the AURAex and AURA2 studies, 1 patient (0.2%) had a prolonged QTc interval that exceeded 500 ms, and 11 patients (2.7%) had a prolonged QTc interval of more than 60 ms from baseline values. A pharmacokinetic analysis of this product predicted a concentration-dependent increase in the incidence of QTc interval prolongation. no arrhythmic events were reported during the AURAex or AURA2 studies (see [Caution]).
Altered Myocardial Contractility
In the AURAex and AURA2 studies (N=411), a decrease in left ventricular ejection fraction (LVEF) of >10% occurred in 2.4% (9/375) of patients with LVEF assessment at baseline and at least 1 follow-up visit and decreased to <50%.
Elderly Patients
Of the patients taking axitinib during the clinical study (N=411), 46% were 65 years of age or older and 13% were 75 years of age or older. A greater number of subjects aged ≥65 years experienced adverse reactions leading to study drug dose adjustment (dose suspension or reduction) compared to subjects aged less (<65 years) (23% vs. 17%). Both categories of patients. Older patients experienced more Grade 3 or higher adverse reactions compared to younger patients (32% vs. 28%).
Reporting of Suspected Adverse ReactionsIt is important to report suspected adverse reactions after a drug has been approved. This action ensures ongoing monitoring of the risk-benefit balance of the product.
[Contraindication]
Hypersensitivity to the active ingredient or any excipients.
This product should not be taken with St. John’s Wort (see [Drug Interactions]).
[Precautions]
EGFR T790MEvaluation of Mutation Status
When considering the use of this product for the treatment of locally advanced or metastatic NSCLC, the status of the EGFR T790M mutation first needs to be clarified. Tumor DNA harvested from tissue samples or circulating tumor DNA (ctDNA) obtained from plasma samples should be tested using a well-validated assay.
When testing for T790M mutation status in tumor DNA (via tissue or plasma samples), a robust, reliable, and sensitive assay must be used.
A positive T790M mutation by tissue or plasma assay suggests treatment with this product. However, if a plasma ctDNA test is used and the result is negative, additional tissue testing should be performed if possible, due to the potential for false negative results with plasma testing.
Interstitial Lung Disease(ILD)
Severe, life-threatening, or fatal interstitial lung disease (ILD) or ILD-like adverse reactions (e.g., noninfectious pneumonia) have been observed in patients using this product in clinical studies. The vast majority of these events improve or resolve after suspension of the drug. Patients with a prior history of ILD, drug-induced ILD, radiation pneumonia requiring steroid hormone therapy, and clinical evidence of active ILD were excluded from clinical studies (see [Adverse Reactions]).
During clinical studies, interstitial lung disease (ILD) or ILD-like adverse reactions (e.g., non-infectious pneumonia) occurred in 2.9% of the 1221 patients treated with this product, with 0.3% of subjects dying. ILD or ILD-like adverse reactions were reported in 11 (2.7%) of 411 patients treated with this product during the two phase II studies, with grade 3 or 4 adverse events in 0.7% and death in 1% of patients. During the study period, ILD occurred in 6.2% of patients of Japanese descent, compared with 1.2% of Asian patients and 2.4% of non-Asian patients (see [Adverse Reactions]).
Carefully examine patients presenting with acute exacerbations and/or unexplained exacerbations of pulmonary symptoms (dyspnea, cough, fever) to rule out ILD. dosing with this product should be suspended while the etiology of these symptoms is sought. If ILD is diagnosed, the product should be permanently discontinued and necessary therapeutic measures should be taken.
Prolonged QTcinterval
Prolonged QTc intervals have been seen in patients taking this product. prolonged QTc intervals can lead to an increased risk of ventricular tachyarrhythmias (such as tip-twist ventricular tachycardia) or sudden death. no arrhythmic events were reported during the AURAex or AURA2 studies (see [Adverse Reactions]). Patients with clinically significant abnormalities in cardiac rhythm or conduction (e.g., QTc interval>470 ms) were excluded from both studies by resting electrocardiogram (ECG) testing (see [Adverse Reactions]).
If possible, avoid this product in patients with congenital long QT interval syndrome. Patients with congestive heart failure, electrolyte abnormalities, or on medications known to prolong the QTc interval should receive regular monitoring of the electrocardiogram (ECG) and electrolytes. Patients with at least two independent ECG tests suggesting a QTc interval >500 ms should be temporarily discontinued until the QTc interval <481 ms or returns to baseline levels (e.g., baseline QTc interval >=481 ms), at which time dosing may be resumed, but should be reduced as per Table 1. Patients with a combination of prolonged QTc interval and any of the following conditions require permanent discontinuation of this product: tip-twisting ventricular tachycardia, polymorphic ventricular tachycardia, signs or symptoms of severe arrhythmias.
Altered Myocardial Contractility
In the AURAex and AURA2 clinical trials, a decrease in left ventricular ejection fraction (LVEF) of >10% occurred in 2.4% (9/375) of patients treated with oseltinib with baseline and at least 1 follow-up LVEF assessment, and decreased to <50%. Based on available clinical trial data, a causal relationship between changes in myocardial contractility and this product cannot be established. Monitoring of cardiac function, including measurement of LVEF function at baseline and during dosing, needs to be considered in patients with known cardiovascular risk and conditions that may affect LVEF. Cardiac monitoring including LVEF function measurement needs to be considered in patients who develop signs and symptoms associated with cardiac events during treatment with this product.
Effect on the ability to drive and operate machinery
This product has no or minimal effect on the ability to drive and operate machinery.
[For pregnant and lactating women]
Female and male contraceptionWomen of childbearing potential should avoid pregnancy while taking this product. Such patients should continue to use effective contraception for at least 2 months for women and at least 4 months for men after completing treatment with this product. The risk of decreased exposure to hormonal contraceptives cannot be ruled out with the combination of this product.
Pregnancy
There are no, or very limited, data on the use of this product in pregnant women. Animal studies suggest reproductive toxicity (embryonic death, embryonic growth retardation, neonatal fetal death, see [Pharmacologic Toxicology]). Based on the mechanism of action and preclinical data, this product may be harmful to the fetus when used in pregnant females. This product should not be used during pregnancy unless the patient’s clinical condition requires treatment with this product.
Lactation.
It is not known whether this product or its metabolites are excreted through human milk. There is also insufficient information to suggest that this product or its metabolites are excreted through the milk of animals. However, the product and its metabolites have been detected in lactating fetuses and have had adverse effects on fetal growth and survival (see [Pharmacology and Toxicology]). Therefore, it cannot be ruled out that this product may have an effect on lactating infants. Therefore, breastfeeding should be discontinued during treatment with this product.
Fertility
There are no data on the effect of this product on fertility in humans. Results from animal studies suggest that this product has effects on the reproductive organs of both females and males and that it impairs fertility (see [Pharmacology and Toxicology]).
[Pediatric Use]
The safety and efficacy of this product in pediatric or adolescent patients younger than 18 years of age is not known. There are no data available on this.
[Geriatric Use]
In clinical trials, 187 of 411 patients (45%) were 65 years of age or older, and 54 patients (13%) were 75 years of age and older. No overall differences in effectiveness were observed based on age. Exploratory analysis showed a higher incidence of grade 3 and 4 adverse reactions in patients aged 65 years and older (32% vs 25%) and more frequent dose adjustments due to adverse reactions (23% vs 17%) compared with patients younger than 65 years.
[Drug interactions
[Drug interactions]
Pharmacokinetic interactions
Potent CYP3A4 inducers may lead to decreased exposure to this product. This product may increase exposure to BCRP substrates.
Active substances that increase plasma concentrations of oxytetracyclineIn vitro studies have confirmed that this product is primarily metabolized via CYP3A4 and CYP3A5 in phase I. In clinical pharmacokinetic studies, coadministration with 200 mg twice-daily itraconazole, a potent CYP3A4 inhibitor, did not produce a clinically significant effect on exposure to this product (a 24% increase in area under the curve (AUC) and a 20% decrease in Cmax). Therefore, CYP3A4 inhibitors are unlikely to have an effect on exposure to this product. Other enzymes that are catalytic to this product have not been identified.
Active substances that reduce plasma concentrations of oseltinibIn clinical pharmacokinetic studies, the combined administration of rifampin (600 mg once daily for 21 days) decreased the steady-state AUC of this product by 78%. Similarly, exposure to the metabolite AZ5104 was reduced, with an 82% and 78% decrease in AUC and Cmax, respectively. It is recommended that concomitant use of this product and strong inducers of CYP3A4 (e.g., phenytoin, rifampin, and carbamazepine) should be avoided. moderate inducers of CYP3A4 (e.g., bosentan, efavirenz, etravirine, and modafinil) may also reduce exposure to this product and should therefore be used with caution and, if possible, should be avoided. When combination of oxitinib with strong inducers of CYP3A is unavoidable, an increase in the dose of oxitinib to 160 mg daily is required. three weeks after discontinuation of strong inducers of CYP3A4, the dose of oxitinib may be restored to 80 mg daily. the combination of this product with St. John’s wort is contraindicated (see [Contraindications]).
Effect of acid-suppressing drugs on ositinibIn clinical pharmacokinetic studies, the combined administration of omeprazole did not have a clinically relevant effect on exposure to this product. This product can be combined with drugs that alter intragastric pH without any restrictions.
Other Active Substances Whose Plasma Concentrations May Be Altered With OcitinibBased on the results of in vitro studies, this product is a competitive inhibitor of the BCRP transporter protein.
In clinical PK studies, the AUC and Cmax of the latter increased by 35% and 72%, respectively, when this product was combined with resulvastatin, a sensitive BCRP substrate. Patients taking this product in combination with drugs that rely on BCRP for distribution and have a narrow therapeutic index should be monitored closely to detect changes in tolerance due to increased exposure to the combined drug in a timely manner.
(See [Pharmacokinetics]).
In clinical PK studies, the combination of this product with simvastatin (a sensitive CYP3A4 substrate) resulted in a 9% and 23% increase in AUC and Cmax, respectively, for the latter. The change was small and therefore unlikely to be clinically significant. It is unlikely that this product interacts with substrates of CYP3A4 in terms of PK. We have not investigated interactions with other enzymes regulated by the pregnane X receptor (PXR) other than CYP3A4. The risk of decreased exposure to hormonal contraceptives cannot be excluded with combined administration of this product.
[Drug Overdose]
During phase I/II clinical studies, a small number of patients had received daily doses of oseltinib up to 240 mg without dose-limiting toxicity. In these studies, patients receiving daily doses of 160 mg and 240 mg of this product had an increase in the frequency and severity of typical EGFR-induced AEs (mainly diarrhea and rash) compared with the 80 mg dose group. However, experience with accidental overdose in humans is more limited. All of these cases were isolated episodic events in which the patient mistakenly took 1 additional dose of the drug without clinical consequences.
There is no specific treatment available after an overdose of this product. If an overdose is suspected, the drug should be suspended and symptomatic treatment administered.
[Clinical Trials]
Two single-arm, open clinical studies were conducted worldwide enrolling patients with EGFR T790M mutation-positive non-small cell lung cancer who had progressed on prior systemic therapy (including an EGFR-TKI) in AURAex (Phase II expansion cohort (n=201)) and AURA2 (n=210). Prior to treatment, all patients were required to be EGFR T790M mutation-positive NSCLC by central laboratory EGFR mutation testing (T790M mutation status of tumor tissue was determined using Roche cobas® in the study). All patients received a once-daily dose of 80 mg of this product. The primary efficacy endpoint for both studies was the objective remission rate (ORR) based on blinded independent center review (BICR) evaluated according to RECIST v1.1. Secondary efficacy endpoints included: duration of remission (DoR), disease control rate (DCR), and progression-free survival (PFS).
Baseline characteristics of the overall study population (AURAex and AURA2) were: median age 63 years; 13% of patients were ≥75 years; female (68%); white (36%); and Asian (60%). All patients received at least one prior therapy. 31% of patients (N=129) had received 1 prior therapy (EGFR-TKI therapy only) and 69% (N=282) had received 2 or more prior therapies. 72% of patients had never smoked, 99% had a World Health Organization (WHO) physical status score of 0 or 1, and 39% had brain metastases (stable for at least 4 weeks and without treatment with corticosteroids). The majority of patients (83%) already had visceral metastases at baseline. the median follow-up time was 6.9 and 6.7 months for the AURAex and AURA2 studies, respectively.
The AURA study (Phase I) was an open, single-arm dose-escalation and expansion Phase I study in which multiple dose-escalation arms included 271 treated patients with locally advanced or metastatic NSCLC. The efficacy and safety of 80 mg once daily was studied in an expanded cohort of 63 treated patients who were EGFR T790M positive by central laboratory testing. Pre-existing therapy included EGFR-TKI and chemotherapy. The demographic characteristics of this T790M-positive study population (n=63) were: median age 60 years; female (62%); white (35%); Asian (59%); patients with World Health Organization (WHO) physical status score 0 or 1 (100%); and nonsmokers (67%). The number of prior lines of treatment ranged from 1 to 9 lines. The median follow-up time was 8.2 months. Table 5 summarizes the efficacy of the AURA study and the pooled analyses of the studies (AURAex and AURA2).
Table5. Efficacy Results of the AURA Study
| I Period | IIIssue | |||
| Efficacy index1 | AURA (Phase I Extended Cohort) (N=63) |
AURAex (II period) (N=201) |
AURA 2 (N=210) |
Summary (N=411) |
| Objective remission rate2,3 % (95% CI) |
62 (48, 74) | 61 (54, 68) | 71 (64, 77) | 66 (61, 71) |
| Duration of Remission (DoR)3 Median value, months (95% CI) |
9.7 (8.3, NE) | NE (NE, NE) | 7.8 (7.1, NE) | NE (8.3, NE) |
| % disease remission beyond 6 months (95% CI) | 72 (54,84) | 83 (74, 89) | 75 (65, 82) | 78 (72, 84) |
| Disease Control Rate (DCR)4 % (95% CI) |
95 (86, 99) | 90 (85, 94) | 91 (87, 95) | 91 (88, 94) |
| Disease progression free survival Median, months (95% CI) |
11 (7, 15) | NE (8.1, NE) | 8.6 (8.3, 9.7) | 9.7 (8.3, NE) |
1 Based on BICR (blinded independent center review), PFS in follow up.
2 Objective remission rates determined by BICR based on RECISTv1.1 for the remission evaluable population (with measurable lesions at baseline according to BICR), pooled for AURA, AURAex, AURA2, and phase II studies, n=60, 199, 199, and 398; NE = not estimable.
3 Only patients who experienced remission were counted; DoR was defined as the time after first documented remission (remission was defined as confirmed complete or partial remission) until documented progression or death in the absence of disease progression.
4 Disease control rate is the percentage of patients in complete or partial remission or with stable disease for ≥6 weeks
Objective remission rates exceeded 50% in all predefined subgroups (number of lines treated, race, age, and region) analyzed.
In the overall population, 86% (227/263 cases) had disease remission at the first imaging scan (6 weeks); 96% (253/263 cases) had disease remission at the 2nd imaging scan (12 weeks).
Clinical studies have not been performed in patients with EGFR T790M de novo mutations.
AURA17 (n=171) is a phase II, open, single-arm study evaluating axitinib (80 mg orally once daily) in patients with locally advanced or metastatic non-small cell lung cancer (NSCLC) (stage IIIB-IV) diagnosed in the Asia-Pacific region, with EGFR-sensitive mutations (EGFRm) and EGFR T790M mutation-positive, previously receiving an already Safety and efficacy in patients with disease progression after prior treatment with approved EGFR-TKI agents. After the appearance and confirmation of disease progression at the time of recent treatment, biopsies need to be performed to allow central laboratory testing for EGFR T790M mutation status (T790M mutation status of tumor tissue was determined using Roche cobas® in the study). The primary aim of validity in this study was the objective remission rate (ORR) as assessed by blinded independent center review (BICR) by RECIST version 1.1. Secondary aims of validity were to assess duration of remission (DoR), disease control rate (DCR), and progression-free survival (PFS).
Baseline characteristics of AURA17 patients were as follows: the majority of patients in this study were female (117/171 [68.4%] patients), Asian (168/171 [98.2%] patients), and Chinese (148/171 [86.5%] patients). The median age of patients at study enrollment was 60.0 years (range: 26 to 82 years), with the largest proportion of patients in the ≥50 and <65 years age group (79/171 [46.2%] patients). 31.6% of patients (N=54) had received 1 prior therapy (EGFR-TKI therapy only), and 68.4% of patients (N=117) had received 2 or more prior therapies. Most patients had metastatic NSCLC (168/171 [98.2%] patients), histologic type adenocarcinoma (165/171 [96.5%] patients) and WHO physical status of 1 (145/171 [84.8%] patients). The mean tumor load at the start of the trial was 66.1 mm (sd, 33.55) based on total target lesion (TL) length diameter at baseline, and the majority of patients had a baseline TL size of 40 to 79 mm (77/171 [45.0%] patients). Most patients had visceral metastases (141/171 [82.5%] patients). The median follow-up time was 4.2 months. Table 6 summarizes the efficacy of the AURA17 study.
Table 6 Efficacy Results of the AURA17 Study
| Efficacy Metrics1 | Overall (N=171) |
China Subgroup (N=148) |
| Objective remission rate2 % (95% CI) |
60.2 (52.4, 67.7) | 59.7 (51.2, 67.8) |
| 89.1 (79.0, 94.5) | 89.9 (81.3, 94.7) | |
| 88.0 (82.0, 92.5) | 88.2 (81.8, 93.0) |
1 Based on BICR (blinded independent center review) in PFS follow-up.
2 Objective remission rates determined by BICR based on RECISTv1.1 for the remission-evaluable population (with measurable lesions at baseline according to BICR) were 166 and 144 for the AURA17 overall and Chinese subgroups, respectively.
3 Only patients who experienced remission were counted; DoR was defined as the time after the first documented remission (remission was defined as confirmed complete or partial remission) until documented progression or death in the absence of disease progression.
4 Disease control rate is the percentage of patients in complete or partial remission or with stable disease for ≥6 weeks.
[Pharmacology and Toxicology]
Pharmacology
Ocitinib is a kinase inhibitor of the epidermal growth factor receptor (EGFR) that binds irreversibly to certain mutants of EGFR (T790M, L858R, and exon 19 deletion) at approximately 9-fold lower concentrations than wild type. In cell culture and animal tumor transplantation tumor models, ositinib had antitumor effects in non-small cell lung cancer cell lines harboring EGFR mutations (T790M/L858R, L858R, T790M/exon 19 deletion, and exon 19 deletion) and weaker antitumor activity against wild-type EGFR gene amplification. After oral administration of oseltinib, two pharmacologically active metabolites (AZ7550 and AZ5104, approximately 10% of the prodrug) were identified in plasma with similar inhibitory profiles as oseltinib. AZ7550 was similar in potency to oseltinib, while AZ5104 had similar potency against EGFR exon 19 deletion and T790M mutation (approximately 8-fold) and wild-type (approximately 15-fold) with stronger activity. In vitro tests showed that at clinical concentrations, ositinib also inhibited HER2, HER3, HER4, ACK1, and BLK activity.
Toxicological studies
Genotoxicity: The results of the oseltinib Ames assay, mouse lymphoma cell assay, and rat in vivo micronucleus assay were all negative.
Reproductive Toxicity: Animal studies have shown that oxitinib may impair fertility in males. Rats and dogs given ositinib for 1 month or longer showed degenerative changes in the testes, and the changes were reversible in rats. In rats given ocitinib at a dose of 40 mg/kg for about 10 weeks, an increase in pre-labour loss was seen after mating between unadministered female rats and administered males at 0.5 times the recommended human dose of AUC at 80 mg, suggesting decreased fertility in males.
Based on the results of animal studies, oxitinib may impair fertility in females. Results of repeated dosing toxicity tests showed that histological changes such as inactivity, degeneration of the corpus luteum in the ovaries, and thinning of the uterine and vaginal epithelium were observed in rats given ocitinib for 1 month or longer when the exposure was 0.3 times the AUC at the recommended human dose of 80 mg. The observed ovarian changes after 1 month of administration were reversible. Female fertility studies showed that ocitinib did not affect the sexual cycle or the number of pregnant animals in female rats given 20 mg/kg/day (approximately 1.5 times the recommended human dose of 80 mg/day Cmax) from two weeks before mating to day 8 of gestation, but it caused early embryonic death. The re-mating of female rats 1 month after discontinuation was reversible.
In a rat embryo/fetus developmental toxicity assay, post-implantation loss and early embryonic death were seen in pregnant rats given oxytetracycline 20 mg/kg/day (plasma exposure approximately 1.5 times the clinical exposure) from before embryo implantation to the end of organogenesis (gestation days 2 to 20). In pregnant rats given ositinib at 1 mg/kg/day or higher (0.1 times the AUC value at the recommended human dose of 80 mg) during the period between implantation and hard palate closure (gestation days 6 to 16), the rate of fetal malformation and variability was suspiciously increased in the administered group compared with the control group.
In a perinatal toxicity test, an increase in total litter abortion and postnatal mortality was seen in pregnant rats given oseltinib 30 mg/kg/day from organogenesis to day 6 of lactation; a slight decrease in mean pup weight at birth and an increase in postnatal mortality were seen at the 20 mg/kg/day dose, and mean pup weight began to increase on days 4-6 of lactation.
Carcinogenicity: No carcinogenicity studies have been conducted with oseltinib.
[Pharmacokinetics]
The pharmacokinetic parameters of this product were characterized in healthy subjects and patients with NSCLC. Based on population pharmacokinetic analysis, the apparent plasma clearance was 14.2 L/h, the apparent volume of distribution was 986 L, and the terminal half-life was approximately 48 hours. The AUC and Cmax of this product were proportional to the dose over the dose range of 20 to 240 mg. Ositinib reached steady state after 15 days of once-daily oral dosing, with approximately 3-fold exposure accumulation. At steady state, circulating plasma concentrations typically remain within a 1.6-fold range during the 24-hour dosing interval.
Absorption
Following oral administration of oseltinib, peak plasma concentrations of oseltinib are reached at a median tmax(min-max) of 6(3-24) hours, with several peaks occurring in the first 24 hours after dosing in some patients. Absolute bioavailability of oxitinib was not measured. Based on a clinical pharmacokinetic study conducted in patients at an 80 mg dose, food does not have a clinically significant effect on the bioavailability of this product. (AUC increased by 6% (90% CI -5, 19) and Cmax decreased by 7% (90% CI -19, 6)). In healthy volunteers taking omeprazole for 5 days and given 80 mg tablets after an increase in intragastric pH, exposure to this product was not significantly affected (AUC and Cmax increased by 7% and 2%, respectively) and the 90% CI for the exposure ratio was within the limit of 80-125%.
DistributionThe mean steady-state volume of distribution (Vss/F) of oxitinib was estimated by population pharmacokinetic modeling to be 986 L, suggesting a broad distribution of the drug within tissues. Plasma protein binding could not be tested due to instability, but may be higher based on the physicochemical properties of the product. Studies have confirmed that this product also binds covalently to rat and human plasma proteins, human serum albumin, and rat and human hepatocytes.
Biotransformation
In vitro studies suggest that oxitinib is mainly metabolized via CYP3A4 and CYP3A5. Of these, CYP3A4-mediated metabolism may be a secondary pathway. In addition, there may be other metabolic pathways that were not fully defined in in vitro studies, and subsequently, two pharmacologically active metabolites (AZ7550 and AZ5104) were detected in preclinical samples as well as in human plasma administered orally with oseltinib; AZ7550 and oseltinib have similar pharmacological properties, while AZ5104 is more potent against both mutant and wild-type EGFR. Following administration of this product, both of these metabolites appeared slowly in plasma with median tmax (min-max) of 24 (4-72) and 24 (6-72) hours, respectively. In human plasma, the prototype ocitinib drug accounted for 0.8% of the total radioactivity and the two metabolites mentioned above accounted for 0.08% and 0.07%, respectively, while most of the radioactivity was covalently bound to plasma proteins. Based on the AUC, the geometric mean of AZ5104 and AZ7550 exposures were approximately 10% of that of oseltinib under steady-state conditions, respectively.
The major metabolic pathways of oseltinib are oxidation and dealkylation. A total of at least 12 components were detected in pooled samples of human urine and feces, of which five components accounted for more than 1% of the total dose. Of these components, the prototype, AZ5104 and AZ7550 accounted for approximately 1.9%, 6.6% and 2.7% of the administered dose, respectively, while a cysteine adduct (M21) and an unknown metabolite (M25) accounted for approximately 1.5% and 1.9%, respectively.
In vitro studies have shown that oxitinib is a competitive inhibitor of CYP 3A4/5, but not of CYP1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6 and 2E1 at clinically meaningful concentrations. Based on in vitro studies, this product is not an inhibitor of UGT1A1 and UGT2B7 in the liver at clinically meaningful concentrations. It may also inhibit UGT1A1 in the intestine, but whether this has clinically relevant effects is unknown.
Elimination
Following single oral administration of this product at a dose of 20 mg, 67.8% of the total dose was collected from stool (1.2% for the prototype drug) and 14.2% of the total dose was collected from urine (0.8% for the prototype drug) by the end of sample collection on day 84. Ocitinib prototypes accounted for approximately 2% of the total elimination, with 0.8% and 1.2% eliminated via urine and feces, respectively.
Interaction with transport proteins
In vitro studies have shown that oxitinib is not a substrate for OATP1B1 and OATP1B3. Also in vitro studies have shown that this product does not inhibit OAT1, OAT3, OATP1B1, OATP1B3 and MATE2K at clinically meaningful concentration conditions. However, it cannot be excluded that this product will interact with MATE1 and OCT2 substrates.
Effect of OsitinibonP-gp andBCRP
In vitro studies have shown that oxitinib is a substrate for P-glycoprotein and breast cancer resistance protein (BCRP), but at clinical doses, oxitinib is unlikely to cause clinically meaningful drug interactions with related active substances. Based on data from in vitro studies, oxitinib is an inhibitor of BCRP and Pgp. However, PXR-regulated enzyme interactions other than CYP3A4 have not been studied (see [Drug Interactions]).
Special PopulationsIn a population pharmacokinetic analysis (n=778), no clinically significant relationships were found between predicted steady-state exposure (AUCss) and the following factors: patient age (range: 21 to 89 years), sex, race (including white, Asian, Japanese, Chinese, and non-Asian non-white), and smoking status (24 current smokers, quitters 232). Population PK analysis suggested that weight was a meaningful covariate, with a -20% to +30% change (95% to 5% quantile) in AUCss for oseltinib in the range of 90 kg to 43 kg compared with AUCss at the median weight (62 kg). When extremes in body weight are taken into account, the range of ratios for the metabolite AZ5104 varies from 11.8% to 9.6% from <43 kg to >90 kg, while the range of AZ7550 ratios varies from 12.8% to 9.9%. The above changes in exposure due to weight differences were not clinically significant.
Hepatic impairment
Ocitinib is primarily eliminated by the liver; therefore, exposure may be increased in patients with hepatic impairment taking this product. No pharmacokinetic studies have been performed in subjects with hepatic impairment. Based on population PK analysis, there was no significant relationship between liver function markers (ALT, AST, and bilirubin) and exposure to axitinib. Serum albumin, a marker of hepatic impairment, had an effect on the PK of ositinib. Clinical studies have been conducted excluding patients with AST or ALT>2.5x upper limit of normal (ULN) or, if due to the malignancy itself, >5.0x ULN or total bilirubin>1.5x ULN. Based on a pharmacokinetic analysis of 44 patients with mild hepatic impairment and 330 patients with normal hepatic function, the exposure was similar in both groups of patients. There are limited data on the use of this product in patients with hepatic impairment (see [DOSAGE]).
Renal Impairment
No pharmacokinetic studies have been conducted in subjects with renal impairment. Based on 330 patients with mild renal impairment (CLcr 60 to <90 mL/min), 149 patients with moderate renal impairment (CLcr 30 to <60 mL/min), 3 patients with severe renal impairment (CLcr 15 to <30 mL/min), and 295 patients with normal renal function (≥90 mL/min) in A population pharmacokinetic analysis of these patients with similar exposures after ocitinib administration. Severe renal impairment may affect the elimination of the drug via the liver. Patients with CLcr ≤ 15 mL/min were not included in the clinical study.
Race.
AURA18 (n=31), a phase I, open study in Chinese patients with locally advanced or metastatic NSCLC with disease progression after prior treatment with an approved marketed EGFR-TKI (with or without other chemotherapy regimens), examined the pharmacokinetic profile of oral oseltinib at two dosing doses (40 mg and 80 mg).
There was slow to moderate and sustained absorption of oxitinib. Increased exposure to ositinib (40 mg to 80 mg) was observed after single and multiple dosing in roughly proportion to the dose administered. Ositinib had low to moderate apparent clearance (14.2 L/hour after a single dose and 15.3 L/hour after multiple doses) and was widely distributed (1113 L).
Ocitinib has a half-life of approximately 40 hours after a single dose and reaches steady-state after 15 days of administration. When steady state (cycle 2, day 1) was reached after multiple doses, the exposure buildup was approximately 3.3-fold, with a flat pharmacokinetic profile at steady state. The two active metabolites, AZ5104 and AZ7550, showed a flat pharmacokinetic profile similar to that of oseltinib at steady state, each cycling at approximately 12% to 15% of oseltinib exposure at steady state.
In comparison to Asian as well as non-Asian patients, Chinese patients showed similar oral oxitinib pharmacokinetic profiles, and oxitinib exposure was not affected by ethnic factors.
[Storage].
Store below 30°C.
[Packaging]
Double aluminum blister packaging, 30 tablets (3 plates) per box.
Double aluminum blister pack, 10 tablets (1 plate) per box.
[Expiration Date].
36 months.
[Executive Standards]
Imported drug registration standard JX20160397.
[Approval Number]
Company Name: AstraZeneca AB
Production Address: Gärtunavägen, SE-151 85 Södertälje, Sweden
Address of China Liaison Office: No. 2 Huangshan Road, Wuxi New District, Jiangsu Province
Zip code: 214028
Quality complaint phone number: 400 828 1755, 800 828 1755
Product Information Toll Free: 400 820 8116, 800 820 8116
Fax: 021-38723255
Website: www.astrazeneca.com.cn