Approval Date: 07/31/2008
Revision date: 09/18/2009; 12/21/2009; 07/18/2011; 09/27/2011.
June 07, 2012; October 30, 2012; February 21, 2013; June 14, 2013; January 06, 2014; June 20, 2014; November 21, 2014; November 24, 2014; 2015 January 20; November 07, 2016; December 20, 2016; February 24, 2017
Varenicline Tartrate Tablets Instructions
Please read the instructions carefully and use under the guidance of your physician.
[Drug Name].
Generic Name: Varenicline Tartrate Tablets
Trade Name: Champix
English Name: Varenicline Tartrate Tablets
Hanyu Pinyin: Jiushisuan Fanikelan Pian
[Ingredients
The main ingredient of this product is varenicline tartrate
Chemical name: 7,8,9,10-tetrahydro-6,10-methylene-6H-pyrazinamide [2,3-h][3]benzazepine-(2R,3R)-2,3-bis Hydroxybutanedioate (1:1)
Chemical structure formula.
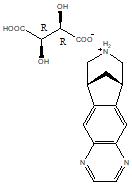
Molecular formula: C13H13N3-C4H6O6
Molecular weight: 361.36
[Properties
This product is a white to off-white film-coated tablet (0.5mg size) or light blue film-coated tablet (1.0mg size), which appears white after removing the coating.
[Indications
This product is indicated for smoking cessation in adults.
[Specifications
0.5mg, 1.0mg
[Dosage].
Dosage
This product is for oral administration. First in 1-week dose increments as follows, after which the recommended dose is 1 mg twice daily.
|
Day 1 to 3: |
0.5mg once daily (white tablets) |
|
Day 4 to Day 7: |
0.5mg twice daily (white tablets) |
|
Day 8 ~ end of treatment: |
1mg twice daily (light blue tablets) |
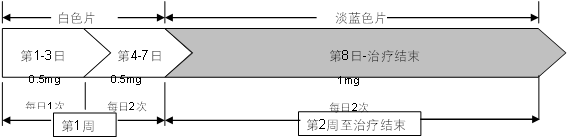
Patients should set a quit date and start taking this product 1 to 2 weeks before this date.
Patients should take this product for 12 weeks of treatment.
For patients who have successfully quit after 12 weeks of treatment, consider renewing for an additional 12-week course at the same dose of 1 mg twice daily to maintain abstinence.
For patients who are unable or unwilling to quit abruptly, a gradual approach to smoking cessation should be considered. Patients should reduce smoking for the first 12 weeks of treatment and quit at the end of the treatment period. Patients should then continue to take this product for an additional 12 weeks to complete a total of 24 weeks of treatment (see [Clinical Trials]).
Patients who are very eager to quit and who have not successfully quit with prior treatment with this product, or who have relapsed after treatment, may benefit from trying again to quit with this product (see [Clinical Trials]).
For patients who cannot tolerate the adverse effects of this product, the dose may be temporarily or chronically reduced to 0.5 mg twice daily.
In smoking cessation treatment, the risk of relapse is elevated in the period immediately following the end of treatment. In patients with a high risk of relapse, medication tapering may be considered. Smoking cessation treatment is more likely to be successful in patients who have a desire to quit and who receive more advice and support.
Patients with renal impairment
Patients with mild (estimated creatinine clearance>50 ml/min and ≤80 ml/min) to moderate (estimated creatinine clearance ≥30 ml/min and ≤50 ml/min) renal impairment do not require dose adjustment.
Patients with moderate renal impairment and intolerable adverse effects may have their dose reduced to 1 mg once daily.
In patients with severe renal impairment (estimated creatinine clearance<30 ml/min), the recommended dose is 1 mg once daily. dosing should be started at 0.5 mg once daily and increased to 1 mg once daily after 3 days. clinical experience with this product in patients with end-stage renal disease is limited, and therefore it is not recommended in this population (see < strong>[Pharmacokinetics]).
Patients with hepatic impairment
Patients with hepatic impairment do not require dose adjustment (see [Pharmacokinetics]).
Elderly Patients
No dose adjustment is required in elderly patients (see [Pharmacokinetics]). Prescribers should consider the renal function status of elderly patients because they are more likely to have decreased renal function.
Patients with sleepwalking disorder
Patients should be instructed to discontinue this product and notify their physician if they develop sleepwalking (see [Precautions]).
Pediatric Patients
The safety and efficacy of this product in children or adolescents under 18 years of age have not been established. The currently available data can be found in [Pharmacokinetics]. This product is not recommended for use in this population.
How to take
This product should be swallowed whole with water and taken before or after meals.
[Adverse Reactions
Smoking cessation itself is associated with a variety of symptoms, whether or not you receive cessation treatment. For example, irritability, depression, insomnia, irritability, frustration, anger, anxiety, difficulty concentrating, restlessness, decreased heart rate, increased appetite, or weight gain have been reported in patients attempting to quit. The design of the clinical study and analysis of the results of this product did not distinguish between the adverse events that occurred and drug or nicotine withdrawal correlations.
Multiple clinical studies of this product have involved approximately 4,000 patients treated for up to 1 year (mean 84 days of dosing). Adverse reactions, if any, usually occurred in the first week of treatment and were mostly mild to moderate in severity. There was no difference in the incidence of adverse reactions by age, race, or sex.
After completing the initial dose escalation, patients took the recommended dose of 1 mg twice daily. the most frequently reported adverse event was nausea (28.6%). Most nausea occurred early in treatment, was mild to moderate in severity, and rarely led to treatment interruption.
The proportion of patients who discontinued treatment due to adverse events was 11.4% in the treatment arm and 9.7% in the placebo arm. Among these patients, the rates of treatment interruption for common adverse events in the treatment group were: nausea (2.7% versus 0.6% in the placebo group), headache (0.6% versus 1.0% in the placebo group), insomnia (1.3% versus 1.2% in the placebo group), and abnormal dreams (0.2% versus 0.2% in the placebo group).
Adverse Reaction Table
The adverse reactions listed in the table below that occurred more frequently in the treatment group than in the placebo group were ranked by system organ type and frequency: very common (≥1/10), common (≥1/100 to <1/10), rare (≥1/1,000 to <1/100), and rare (≥1/10,000 to <1/1,000)). Adverse reactions that occurred with similar frequency are listed in order of increasing severity. All adverse drug reactions (ADRs) listed in the table below are based on the assessment of data from pre-marketing Phase 2-3 studies and updated based on pooled data from 18 placebo-controlled pre-marketing and post-marketing studies, which included approximately 5,000 patients treated with varenicline. Reported post-marketing adverse reactions are also included at frequencies unknown (not estimable from the available data). Within each frequency group, adverse reactions are listed in order of severity from highest to lowest.
|
System organ classification |
Adverse Drug Reactions |
|
< strong>Infection and Infestation |
|
|
Very common |
Nasopharyngitis |
|
Common |
Bronchitis, sinusitis |
|
Rarely |
Fungal infections, viral infections |
|
Abnormalities of the blood and lymphatic system |
|
|
Rare |
Decreased platelet count |
|
Metabolic and nutritional abnormalities |
|
|
Weight gain, loss of appetite, increased appetite |
|
|
Rare |
Bothered by thirst |
|
unknown |
Diabetes, hyperglycemia |
|
Psychiatric abnormalities |
|
|
Very common |
Abnormal dreams, insomnia |
|
Rarely seen |
Panic reaction, abnormal thinking, fidgeting, mood swings, depression*, anxiety*, hallucinations*, increased sexual desire, decreased sexual desire |
|
Rare |
Irritable, slow thinking |
|
unknown |
Suicide ideation, psychosis, aggression, abnormal behavior, sleepwalking disorder |
|
Neurological abnormalities |
|
|
Very common |
Headaches |
|
Common |
Drowsiness, dizziness, taste disorders |
|
Rarely seen |
Seizures, tremors, lethargy, hyperalgesia |
|
Rare |
Cerebrovascular accidents, hypertonia, dysarthria, ataxia, hypoesthesia, circadian sleep rhythm disturbances |
|
Eye abnormalities |
|
|
Rarely seen |
Conjunctivitis, eye pain |
|
Rare |
Dark spots, scleral decoloration, dilated pupils, photophobia, myopia, tearfulness |
|
Ear and vagal abnormalities |
|
|
Rarely seen |
Tinnitus |
|
Cardiac abnormalities |
|
|
Rarely |
Angina pectoris, tachycardia, palpitations, rapid heart rate |
|
Rare |
Atrial fibrillation, ECG ST-segment depression, ECG T-wave amplitude depression |
|
unknown |
myocardial infarction |
|
Vascular abnormalities |
|
|
rare |
Increased blood pressure, hot flashes |
|
Respiratory, thoracic and mediastinal abnormalities |
|
|
Common |
Hard to breathe, coughing |
|
Rarely |
Upper airway inflammation, airway congestion, vocal difficulties, allergic rhinitis, throat irritation, sinus congestion, upper airway cough syndrome, rhinorrhea |
|
Sore throat, snoring |
|
|
Gastrointestinal abnormalities |
|
|
Very common |
Nasty |
|
Common |
Gastroesophageal reflux disease, vomiting, constipation, diarrhea, bloating, abdominal pain, toothache, indigestion, flatulence, dry mouth |
|
Rarely seen |
Blood in stool, gastritis, change in bowel habits, belching, ulcerative stomatitis, gum pain |
|
Rare |
Vomiting blood, abnormal stools, and Thick and greasy tongue |
|
Dermal and subcutaneous tissue abnormalities |
|
|
Common |
Rash, itching |
|
Rarely |
Redness, acne, hyperhidrosis, night sweats |
|
unknown |
Severe skin reactions include Stevens-Johnson syndrome, erythema multiforme, angioneurotic edema |
|
Musculoskeletal and connective tissue abnormalities |
|
|
Common |
Joint pain, myalgia, back pain |
|
Rarely |
Muscle spasms, musculoskeletal pain in the chest |
|
Rare |
Joint stiffness, costochondritis |
|
Kidney and urinary abnormalities< |
|
|
Rarely seen |
Frequent urination, nocturia |
|
Rare |
sugaruria, polyuria |
|
Reproductive system and breast abnormalities |
|
|
Rarely seen |
Menstrual cramps |
|
Rare |
Vaginal discharge, sexual dysfunction |
|
Systemic abnormalities and dosing site abnormalities |
|
|
Common |
chest pain, fatigue |
| < p>Rarely seen |
Chest discomfort, flu-like illness, fever, malaise, discomfort |
|
Rare |
Cold sensation, cysts |
| < p>Check | |
|
Common |
Abnormal liver function tests |
|
Rarely seen |
Abnormal semen analysis, elevated C-reactive protein, decreased blood calcium, |
Postmarketing experience.
The following adverse events were reported during the post-marketing use of this product. Because these events were spontaneously reported by a population of uncertain size, it was not possible to estimate the incidence of the events or to establish a causal relationship with the drug.
In patients taking this product for smoking cessation, reported events included depression, mania, psychosis, hallucinations, paranoia, delusions, homicidal ideation, aggression, hostility, anxiety, and panic, and suicidal ideation, suicide attempts, and completed suicide (see [Precautions]). With or without pharmacotherapy for smoking cessation, patients may experience nicotine withdrawal symptoms and exacerbation of pre-existing psychiatric disorders. Not all patients understand whether they have a psychiatric disorder, and not all patients are no longer smoking.
Post-marketing reports of new onset or worsening of pre-existing epilepsy have been reported in patients taking this product for treatment (see [Precautions]).
Postmarketing reports of increased alcohol euphoria in patients while taking varenicline. Some patients reported neuropsychiatric symptoms, including abnormal behavior and sometimes aggressive behavior (see [Precautions]).
There are postmarketing reports of hypersensitivity reactions, including angioneurotic edema, in patients treated with varenicline (see [Precautions]).
There are postmarketing reports of rare but serious skin reactions in patients taking varenicline, including Stevens-Johnson syndrome and erythema multiforme (see [Precautions]).
There have been postmarketing reports of myocardial infarction (MI) and cerebrovascular accidents (CVA) including ischemic and hemorrhagic events in patients treated with varenicline. In the majority of such reported events, patients had prior cardiovascular system disease and/or other risk factors. Although smoking is a risk factor for myocardial infarction and cerebrovascular accident, varenicline cannot yet be excluded based on a temporal correlation between varenicline use and events (see [Cautions]).
There have been reports of hyperglycemia in patients taking varenicline.
Sleepwalking has been reported in patients treated with varenicline, leading in some cases to behaviors harmful to self, others, or property (see [precautions]).
[Contraindications
People with hypersensitivity to the active ingredient or any of the excipient ingredients of this product.
Warning
1. Neuropsychiatric symptoms and suicide
In post-marketing experience with this product, there have been reports of behavioral or thought changes, anxiety, psychosis, mood changes, aggressive behavior, depression, suicidal ideation and behavior, and suicide attempts.
A large randomized, double-blind, active and placebo-controlled study compared the risk of serious psychoneurological events with varenicline, bupropion, nicotine replacement therapy patch (NRT), or placebo for smoking cessation in patients with and without a history of psychiatric illness. The primary safety endpoint was a composite of psychoneurological adverse events reported in the postmarketing experience.
Compared with the primary composite endpoint in the placebo group, the use of varenicline in patients with and without a history of psychiatric illness did not increase the risk of serious psychoneurological adverse events (see [clinical trial]-for subjects with and without a Psychoneurological Safety Study in Subjects with and without a History of Psychiatric Disorders).
Depressed mood may be a nicotine withdrawal symptom. Depression, including rare suicidal ideation and suicide attempts.
Clinicians should be aware of the potential for severe psychoneurologic symptom emergencies in patients who are attempting to quit with or without treatment. If a patient develops severe psychoneurologic symptoms while on varenicline therapy, the patient should immediately discontinue varenicline and contact a health care provider for reassessment of therapy.
History of psychiatric illness
With or without medication, smoking cessation itself is associated with worsening of an underlying psychiatric illness, such as depression.
The varenicline smoking cessation study provided data related to patients with a history of mental illness (see [Clinical Trials]).
In smoking cessation clinical trials, patients with a history of psychiatric disorders reported a higher frequency of psychoneurological adverse events compared with patients without a history of psychiatric disorders, regardless of treatment (see [Clinical Trials]).
Patients with a history of psychiatric disorders should be given special attention and informed.
Analysis of clinical studies
A meta-analysis of 5 randomized, double-blind, placebo-controlled studies including 1907 patients (1130 in the varenicline group and 777 in the placebo group) assessed suicidal ideation and behavior using the Columbia Suicide Severity Rating Scale (C-SSRS). The meta-analysis included one study of patients with a history of schizophrenia or schizoaffective disorder (N=127) and another study of patients with a history of depression (N=525). The results showed that the incidence of suicidal ideation and/or behavior was not increased in patients treated with varenicline compared with those treated with placebo, with a risk ratio (RR) of 0.79 (95% confidence interval [CI]: 0.46, 1.36), as shown in Table 1. Of the 55 patients who reported suicidal ideation or behavior, 48 (24 in the varenicline group and 24 in the placebo group) were from two trials that included patients with a history of schizophrenia, schizoaffective disorder, or depression. Fewer events were observed in the other three trials (4 in the varenicline group and 3 in the placebo group).
Table 1. Number and risk ratio of patients with suicidal ideation and/or behavior as assessed by the C-SSRS in a meta-analysis of 5 clinical studies comparing varenicline and placebo
|
Vaniklang (N=1130) |
Placebo (N=777) |
|
|
Patients with suicidal ideation and/or behavior*[n (%)]** |
28 (2.5) |
27 (3.5) |
|
Exposure (patient years) |
325 |
217 |
|
Risk ratio# (RR; 95% CI) |
0.79 (0.46, 1.36) |
|
|
* In these events, one patient in each treatment group reported suicidal behavior ** Patients with an event within 30 days of treatment; % not weighted according to study # RR per 100 patient-years |
||
A meta-analysis of 18 double-blind, randomized, placebo-controlled clinical studies (including the 5 studies that collected the C-SSRS described in Table 1) was conducted to assess the psychoneurological safety of varenicline. This pooled analysis included 8521 patients (5072 in the varenicline group and 3449 in the placebo group), some of whom had psychiatric disorders at baseline. The results showed that patients in the varenicline and placebo treatment groups had similar rates of total psychoneurological adverse events (excluding insomnia) with a risk ratio (RR) of 1.01 (95% CI: 0.891.15). Pooled data from these 18 trials showed that patients in the varenicline group had similar rates of all types of adverse psychiatric events to those in the placebo group. Table 2 describes the most frequently reported (≥1%) categories of adverse events related to psychiatric safety (excluding insomnia and sleep disturbances).
Table 2. Psychiatric Adverse Events Occurring in ≥1% of Patients in a Pooled Analysis of 18 Clinical Studies
|
Vaniklang (N=5072) |
Placebo (N=3449) |
|
|
Anxiety disorders and symptoms |
253 (5.0) |
206 (6.0) |
|
Depressive mood disorders and disturbances |
179 (3.5) |
108 (3.1) |
|
Mood disorders and disorders NEC* |
116 (2.3) |
53 (1.5) |
|
* NEC=Not Elsewhere Classified Count (percentage) equals number of patients reporting events |
||
Observational studies
Four observational studies, each containing 10,000 to 30,000 varenicline users with adjusted analyses, compared the risk of serious neuropsychiatric symptoms (including hospitalization for neuropsychiatric disorders, fatal and nonfatal self-harm) between varenicline users and users prescribed NRT or bupropion. All studies were retrospective cohort studies that included patients with and without a history of psychiatric disorders. All studies used statistical methods to control for confounding factors (including preferential prescribing of varenicline to healthier patients), but residual confounding may still exist.
Two of the studies found no difference in the risk of hospitalization for neuropsychiatric disorders between varenicline users and nicotine patch users (hazard ratio [HR] 1.14; 95% confidence interval [CI]: 0.56-2.34 in the first study; hazard ratio 0.76 in the second study; 95% CI. 0.40-1.46). However, neither study validated the diagnostic codes used to determine outcomes based on medical records. The test efficacy required to detect differences between these two studies was insufficient. The third study reported no difference between varenicline users and bupropion users in the risk of psychiatric adverse events diagnosed during emergency department visits or hospitalizations (HR 0.85; 95% CI: 0.55-1.30). Bupropion was also associated with neuropsychiatric adverse events. A fourth study showed no evidence of a higher risk of fatal and nonfatal self-harm in patients using varenicline compared with those using NRT (HR 0.88; 95% CI: 0.521.49). Although detectable suicides that occurred during the three months after patients started any drug therapy were relatively rare (two cases in 31,260 varenicline users and six cases in 81,545 NRT users), the study also had serious limitations. Most importantly, these data were collected after reports of neuropsychiatric adverse events in varenicline users became known to the public. Varenicline users presented with fewer comorbid conditions that might put them at risk for neuropsychiatric adverse events, suggesting that patients with a history of neuropsychiatric disorders were prescribed NRT preferentially and healthier patients were prescribed varenicline preferentially.
The results examined in these studies did not include the full range of reported neuropsychiatric adverse events.
2 Epilepsy
Seizures have been reported in patients treated with varenicline in clinical studies and post-marketing experience. Some patients had no history of epilepsy, while others had a distant or well-controlled history of epilepsy. In most cases, seizures occurred within the first month of treatment. This potential risk should be weighed against the benefit before prescribing varenicline to patients with a history of epilepsy or other factors that may lower the seizure threshold. If seizures occur during treatment, patients are advised to discontinue varenicline and contact their healthcare provider immediately (see [Adverse Reactions]).
3 Interactions with Alcohol
Post-marketing reports of increased alcohol euphoria in patients while taking varenicline have been reported. A causal relationship between these events and the administration of varenicline has not been confirmed. Some cases described abnormal and sometimes aggressive behavior, and the events were often accompanied by memory loss. While taking varenicline, patients are advised to reduce alcohol consumption without knowing whether varenicline may have an effect on their alcohol tolerance (see [Adverse Reactions]).
4 Accidental injury
Post-marketing reports of traffic accidents, attempted traffic accidents, or other unintentional injuries have been reported in patients taking this product. In some cases patients have reported drowsiness, dizziness, loss of consciousness, or difficulty concentrating while driving or operating machinery, which can lead to injury or cause concern that an injury may result. Patients should be advised to exercise caution when driving, operating machinery, or other potentially hazardous activities without knowing how taking this product may affect them.
5. Cardiovascular Events
This product was studied in a placebo-controlled clinical study in patients with stable cardiovascular disease in approximately 350 patients per treatment group. All-cause and cardiovascular mortality was lower in the varenicline treatment group than in the placebo group in the study, but the incidence of certain nonfatal cardiovascular events was higher than in the placebo group (see [Clinical Trials]). The table below shows the incidence of fatal events and selective non-fatal serious cardiovascular events in the varenicline group that were higher than in the placebo group. These events were adjudicated by an independent blinded committee. The incidence of non-fatal serious cardiovascular events that occurred at similar rates in both groups or were more common in the placebo group are not included. In each row, patients with multiple identical cardiovascular events were recorded only 1 time. Some of the measures performed in patients requiring coronary revascularization were performed as part of non-fatal myocardial infarction and hospitalization for angina pectoris.
Table 3 Mortality and adjudicated nonfatal serious cardiovascular events in clinical studies of varenicline tartrate tablets versus placebo control in patients with stable coronary heart disease
|
Mortality and cardiovascular events |
Vanikram group (N=353) n (%) |
Placebo group (N=350) n (%) |
|
|
Mortality (52 weeks cardiovascular or all-cause)) |
|||
|
Cardiovascular death |
1 (0.3) |
2 (0.6) |
|
|
2 (0.6) |
|||
|
All-cause mortality |
2 (0.6) |
5 (1.4) |
|
|
Non-fatal cardiovascular events (ratio Varenicline group > placebo group) |
|||
|
< span style="text-decoration:underline">within 30 days of treatment |
|||
|
Non-fatal myocardial infarction |
4 (1.1) |
1 (0.3) |
|
|
Non-fatal stroke |
2 ( 0.6) |
0 (0) |
|
|
30 days to 52 weeks post-treatment |
|||
|
Non-fatal myocardial infarction |
3 (0.8) |
2 (0.6) |
|
|
Requiring coronary artery reconstruction |
7 (2.0) |
2 (0.6) |
|
|
Hospitalized for angina |
6 (1.7) |
6 (1.7) |
4 (1.1) |
|
Transient ischemic attack |
1 (0.3) |
0 (0) |
|
|
Newly diagnosed peripheral vascular disease (PVD) or hospitalization for peripheral vascular disease |
5 (1.4) |
2 (0.6) |
|
A systematic evaluation of the cardiovascular safety of varenicline tartrate was performed in a meta-analysis including 15 clinical trials with a treatment duration of ≥12 weeks and a total of 7002 patients (4190 in the varenicline group and 2812 in the placebo group). The above-mentioned clinical studies in patients with stable cardiovascular disease were included in the meta-analysis. All-cause death (6 [0.14%] in the varenicline group and 7 [0.25%] in the placebo group) and cardiovascular death (2 [0.05%] in the varenicline group and 2 [0.07%] in the placebo group) occurred at lower rates in the varenicline group than in the placebo group as a result of the meta-analysis.
The critical cardiovascular safety analysis included the occurrence and timing of the composite end point of major adverse cardiovascular events (MACE), defined as cardiovascular death, nonfatal myocardial infarction, and nonfatal stroke. These events included in the endpoints were adjudicated by an independent blinded committee. In general, MACE occurred less frequently in the clinical trials included in the meta-analysis, as shown in the table below. These events occurred primarily in patients with known cardiovascular disease.
Table 4 Number of MACE cases, risk ratios, and ratio differences in the meta-analysis of the 15 clinical studies that included varenicline compared with placebo
|
Vanikram group N=4190 |
Placebo group N=2812 |
|
|
Number of MACE cases, n (%) |
13 (0.31%) |
6 (0.21%) |
|
Patients – Year of Exposure |
1316 |
839 |
|
Risk Ratio (95% CI) |
||
|
1.95 (0.79, 4.82) |
||
|
Ratio difference per 1,000 patient-years (95% CI) |
||
|
6.30 (-2.40, 15.10) |
||
*Including MACE occurring after 30 days of treatment
This product has not been studied in patients with unstable cardiovascular disease or in patients who developed cardiovascular disease within 2 months prior to screening. Patients are advised to contact their healthcare provider if they develop new or worsening symptoms of cardiovascular disease. The benefit-risk ratio should be weighed when using this product in smokers with cardiovascular disease. Smoking is an independent and important risk factor for cardiovascular disease. This product has been shown to be effective in increasing the odds of quitting smoking for up to 1 year compared with placebo.
6. Sleepwalking disorder
Sleepwalking has been reported in patients using varenicline. Some cases have described harmful behavior toward self, others, or property. Patients should be advised to discontinue varenicline and notify their physician if they experience sleepwalking (see [Adverse Reactions]). .
General Precautions
1. Angioneurotic edema and hypersensitivity reactions
Postmarketing reports of hypersensitivity reactions, including angioneurotic edema, have been reported in patients treated with this product (see [Adverse Reactions]). Clinical signs include swelling of the face, mouth (tongue, lips, gums), extremities, and neck (pharynx and larynx). In addition, there have been rare reports of life-threatening angioneurotic edema requiring emergency medical management due to resulting respiratory dysfunction. Patients should be advised to discontinue the product and seek immediate medical attention in the event of these symptoms.
2. Severe skin reactions
There have been postmarketing reports of rare but serious skin reactions in patients taking this product, including Stevens-Johnson syndrome and erythema multiforme [see [Adverse Reactions]]. Because these skin reactions can be life-threatening, patients should be advised to stop taking the product and contact their healthcare provider as soon as they develop a rash with mucosal lesions or any other signs of hypersensitivity reactions.
3. Nausea
Nausea is the most common adverse reaction in the treatment of this product. Nausea is usually mild to moderate and transient, but for some patients, nausea can persist for several months. The incidence of nausea is related to the dose administered. Initial dose titration is beneficial in reducing the incidence of nausea. When the maximum recommended dose of 1 mg twice daily was given to patients after a gradual increase in dose, the incidence of nausea was 30%, compared to 10% in the placebo control group. The incidence of nausea was 16% in the placebo group and 11% in the placebo group when varenicline was given to patients at 0.5 mg twice daily after a gradual dose increase in the starting phase. In the 12-week study in which patients were given 1 mg of varenicline twice daily, approximately 3% of patients discontinued treatment due to nausea. Dose reduction is recommended if the patient cannot tolerate it.
4. Drug abuse and dependence
Less than 1 in 1000 patients reported euphoria in clinical studies of this product. Higher doses (>2 mg) of this product are more likely to cause gastrointestinal distress, such as nausea and vomiting. No evidence was found in clinical studies that increasing doses were required to maintain therapeutic efficacy, suggesting that this product is not tolerated. When abruptly discontinued, irritability and sleep disturbances occur in no more than 3% of patients. This suggests that in some patients, varenicline may produce mild somatic dependence, but is not associated with addiction.
In a laboratory study of human propensity to abuse, a single oral dose of 1 mg of varenicline did not produce a significant positive or negative subjective response in smokers. In nonsmokers, 1 mg varenicline produced some increase in positive subjective responses, but was accompanied by an increase in negative adverse effects, particularly nausea. A single oral dose of 3 mg varenicline produced an uncomfortable subjective response in both smokers and nonsmokers.
[For pregnant and lactating women].
Pregnant women
Risk Summary
There are insufficient available human data on the use of varenicline in pregnant women to provide information on the risks associated with the drug. Smoking during pregnancy poses risks to the mother, fetus, and newborn (see Clinical Considerations). In animal studies, varenicline did not cause severe malformations when administered during the organogenesis phase at exposures equivalent to 50 times the maximum recommended human dose (MRHD), but resulted in reduced fetal weight in rabbits. In addition, maternal exposure of varenicline administered to pregnant rats during organogenesis through lactation at a maternal exposure equivalent to 36 times the human exposure at MRHD produced developmental toxicity in the offspring (see data).
The predicted background risk of cleft lip and palate in infants born to pregnant women who smoked during gestation was increased by approximately 30% compared with nonsmoking pregnant women. For the referred population, the background risk for other major birth defects and miscarriages is unknown. In the general US population, the predicted background risks for clinically confirmed major birth defects and miscarriage in pregnancy are 2-4% and 15-20%, respectively.
Clinical Considerations
Maternal and/or embryonic/fetal risks associated with disease
Smoking during pregnancy results in an increased risk of cleft lip and palate, premature rupture of membranes, placenta praevia, placental abruption, ectopic pregnancy, fetal growth restriction and low birth weight, stillbirth, preterm delivery and gestational shortening, neonatal death, sudden infant death syndrome, and infant pulmonary hypoplasia. It is not known whether smoking cessation with varenicline during pregnancy reduces these risks.
Data
Animal data
Pregnant rats and rabbits were given oral doses of varenicline succinate up to 15 and 30 mg/kg/day, respectively, during organogenesis. Although no fetal structural abnormalities were observed in either animal, maternal toxicity (as evidenced by reduced body weight gain) and fetal weight loss were seen in pregnant rabbits given high doses of varenicline (50 times the maximum recommended human dose of 1 mg twice daily exposure based on AUC); no fetal weight loss was seen in rabbits exposed at 23 times the maximum recommended human dose (based on AUC).
In a prenatal and postnatal developmental study, pregnant rats received 15 mg/kg/day of oral varenicline succinate from fetal organogenesis to lactation. Maternal toxicity (as evidenced by reduced body weight gain) was observed at a dose of 15 mg/kg/day (equivalent to 36 times the exposure at the maximum recommended human dose, based on AUC). However, at the highest maternal dose of 15 mg/kg/day, the offspring showed reduced fertility and increased auditory startle response.
Lactation
Risk summary
No information is available on the presence of varenicline in breast milk, its effect on breastfed infants, or its effect on lactation. In animal studies, varenicline was present in the milk of lactating rats (see data). However, animal data cannot reliably predict drug levels in breast milk due to differences in lactation physiology between species. The lack of clinical data during lactation makes it impossible to definitively determine the risk of varenicline to the nursing infant; however, the developmental and health benefits of breastfeeding, the maternal clinical need for varenicline, and any potential adverse effects of varenicline or the underlying maternal condition on the breastfed infant should be considered together.
Clinical Considerations
Because there are no data on the presence of varenicline in breast milk and the effects on breastfed infants, breastfeeding women should monitor their infants for seizures and excessive vomiting, which are adverse reactions that have been seen in adults and may be clinically relevant in breastfed infants.
Data
In a prenatal and postnatal developmental study, pregnant rats were given up to 15 mg/kg/day of oral varenicline succinate from gestation to lactation, and the mean serum concentration of varenicline in the lactated pups was 5-22% of the maternal serum concentration.
Fertility
There are no clinical data on the effect of this product on fertility.
Results from male and female fertility studies in rats suggest that this product may not be harmful to human fertility (see [Pharmacology and Toxicology]).
[Pediatric use
See [Dosage].
[Geriatric Use
See [Dosage].
[Drug Interactions].
Based on the properties of varenicline and current clinical experience, no clinically meaningful interactions have been identified between this product and other drugs. No dose adjustment of this product and the following combination drugs is required.
In vitro studies have shown that varenicline is unlikely to alter pharmacokinetic parameters for compounds metabolized primarily by cytochrome P450; since less than 10% of varenicline is cleared by metabolism, the active substances known to affect the cytochrome P450 system are unlikely to affect the pharmacokinetic parameters of varenicline (see [Pharmacokinetics]) and therefore no dose adjustment of this product is required.
In vitro studies have shown that therapeutic concentrations of varenicline have no inhibitory effect on human renal transport proteins. Therefore varenicline is unlikely to affect active substances cleared by renal secretion (e.g., metformin-as shown below).
Metformin: Varenicline does not affect the pharmacokinetic parameters of metformin. Metformin also does not affect the pharmacokinetic parameters of varenicline.
Cimetidine: Concomitant administration of varenicline and cimetidine decreased the renal clearance of varenicline and increased its systemic exposure by 29%. No dose adjustment was required for concomitant application of both drugs in subjects with normal renal function or in patients with mild or moderate renal impairment. In patients with severe renal impairment, concomitant application of both drugs should be avoided.
Digoxin: Varenicline does not alter the steady-state pharmacokinetic parameters of digoxin.
Warfarin: Varenicline does not alter the pharmacokinetic parameters of warfarin. Prothrombin time (measured as INR) is not affected by varenicline. Smoking cessation itself may alter the pharmacokinetic parameters of warfarin.
Alcohol: Clinical information on the potential interaction of alcohol with varenicline is limited.
Concomitant use with other smoking cessation treatments.
Bupropion: Varenicline does not alter the steady-state pharmacokinetic parameters of bupropion.
Nicotine replacement therapy (NRT): Varenicline was given concomitantly with transdermal NRT to smokers for 12 days, and the mean systolic blood pressure detected on the last day of the study was significantly lower (mean 2.6 mm Hg), a change that was statistically significant. In this study, the incidence of nausea, headache, vomiting, dizziness, dyspepsia, and fatigue was higher in the combined treatment group than in the group treated with NRT alone.
The safety and efficacy of this product in combination with other smoking cessation therapies has not been studied.
[Overdose].
Overdose has not been reported in premarketing clinical studies.
In the event of an overdose, standard supportive therapy should be given as required.
Studies have shown that varenicline can be cleared by dialysis in patients with end-stage renal disease (see [Pharmacokinetics]), but there is no experience with dialysis for drug overdose.
[Clinical Trials].
Clinical Efficacy and Safety
Three clinical studies in long-term smokers (≥10 cigarettes/day) demonstrated the effectiveness of this product for smoking cessation. 2619 subjects were treated with 1 mg twice daily (with dose escalation during the first week), 669 subjects were treated with bupropion at 150 mg twice daily (also with dose escalation), and 684 subjects were treated with bupropion at 150 mg twice daily (also with dose escalation). increments), and 684 subjects received placebo treatment.
Controlled clinical study
Two identically designed prospective double-blind clinical studies compared the smoking cessation effects of this product (1 mg twice daily), bupropion extended-release (150 mg twice daily), and placebo. In the 52-week study, patients received 12 weeks of treatment followed by a 40-week treatment-free phase.
The primary endpoint in both studies was the 4-week sustained abstinence rate (4W-CQR) at weeks 9 to 12, as confirmed by carbon monoxide (CO) testing. The results for the primary endpoint showed superiority and statistical significance for this product over bupropion and placebo.
A key secondary endpoint for both studies was the sustained abstinence rate (CA) at week 52 after a 40-week treatment-free phase. The sustained abstinence rate was defined as the proportion of all subjects who did not smoke (not a puff) and whose exhaled breath carbon monoxide measurement was not greater than 10 ppm between weeks 9 and 52. The 4-week sustained abstinence rates (9 to 12 weeks) and sustained abstinence rates (9 to 52 weeks) for Study 1 and Study 2 are shown in the following table.
|
Study 1 (n=1022) |
Study 2 (n=1023) |
|||
|
4W CQR |
CA Wk 9 to 52 |
4W CQR |
CA Wk 9 to 52 |
|
|
Varenicline Tartrate Tablets |
44.4% |
22.1% |
44.0% |
23.0% |
|
Bupropion |
29.5% |
16.4% |
30.0% |
15.0% |
|
Placebo |
< p style="text-align: center">17.7% |
8.4% |
17.7% |
10.3% |
|
Ratio Varenicline tartrate tablets vs. placebo |
3.91 p<0.0001 |
3.13 p<0.0001 |
3.85 p<0.0001 |
2.66 p<0.0001 |
|
Ratio Varenicline tartrate tablets vs. bupropion |
1.96 p<0.0001 |
1.45 p=0.0640 |
1.89 p<0.0001 |
1.72 p=0.0062 |
Patients’ reported smoking cravings, withdrawal symptoms, and reinforcing effects of smoking
Subjects randomized to the treatment group in Study 1 and the pharmacotherapy phase of Study 2 had significantly reduced smoking cravings and withdrawal symptoms compared with the placebo group. The reinforcing effect allowed for the persistence of smoking behavior in patients during treatment, and it also significantly reduced the reinforcing effect of smoking compared with placebo. The effects of varenicline on smoking craving, withdrawal symptoms, and smoking reinforcement were not evaluated during the treatment-free long-term follow-up phase.
Status Maintenance of Smoking Cessation Study
The third study evaluated the benefit of a follow-up 12-week treatment for maintenance of abstinence status. Patients (n=1,927) first received 12 weeks of open treatment with 1 mg twice daily, and at the end of week 12, those who stopped smoking were then randomized to either continue taking 1 mg twice daily or placebo for 12 weeks, for a total study period of 52 weeks.
The primary endpoint of the study was the rate of sustained abstinence (CA) confirmed by carbon monoxide testing at weeks 13 to 24 of the double-blind treatment phase. The key secondary endpoint was the sustained withdrawal rate (CA) at weeks 13 to 52.
The study showed that the subsequent 12 weeks of varenicline treatment (1 mg twice daily) facilitated maintenance of abstinence, with a significant difference compared with taking placebo; the sustained abstinence advantage of this product over placebo was maintained through week 52. The primary results are summarized in the following table.
Continued abstinence rates in patients treated with varenicline and placebo
|
Varenicline Tartrate Tablets n=602 |
Placebo n=604 |
Differences (95% confidence interval) |
Ratio (95% confidence interval) |
|
|
CA* wk 13-24 |
70.6% |
49.8% |
20.8% (15.4%, 26.2%) |
2.47 (1.95, 3.15) |
|
CA* wk 13-52 |
44.0% |
37.1% |
6.9% (1.4%, 12.5%) |
1.35 (1.07, 1.70) |
*CA : Continuous abstinence rate
There is limited clinical experience with this product in blacks, and its efficacy in this population has not been established.
Flexibility in setting quit days between week 1 and week 5
The efficacy and safety of varenicline was evaluated in smokers who had a flexible quit date between week 1 and week 5. In this 24-week study, patients received 12 weeks of treatment followed by 12 weeks of treatment-free follow-up. The 4-week sustained abstinence rate (4W CQR) from week 9 to week 12 was 53.9% for the varenicline group and 19.4% for the placebo group (difference = 34.5%, 95% confidence interval: 27.0% to 42.0%) and the sustained abstinence (CA) from week 9 to week 24 was 35.2% for the varenicline group and 12.7% for the placebo group (difference = 22.5%, 95% confidence interval: 15.5%). 95% confidence interval: 15.8% to 29.1%). Patients who are unwilling or unable to set a quit day on their own within 1 to 2 weeks may first receive treatment and set a quit day within 5 weeks.
Study in patients retreated with this product
In a double-blind placebo-controlled trial, this product was evaluated in 494 patients who had tried to quit smoking with this product but either failed to quit, or relapsed after treatment. The trial excluded from participation those patients who had concerns about adverse events on prior treatment. Patients were randomly assigned in a 1:1 ratio to either the Benadryl group (1 mg twice daily) (n=249) or the placebo group (n=245) to receive 12 weeks of treatment and to receive up to 40 weeks of post-treatment follow-up. Patients recruited for this study had attempted to take this product to quit smoking at least three months prior to entry into this study (total treatment duration of at least two weeks) and had smoked for at least four weeks thereafter.
A higher abstinence rate from week 9 to week 12 and from week 9 to week 52 was confirmed by carbon monoxide testing in the treatment group compared to the placebo group. The primary results are summarized in the following table.
Continued abstinence rates in patients treated with varenicline and placebo
|
This product n=249 |
Placebo N=245 |
ratio (95% confidence interval) p-value |
|
|
CA* wk 9-12 |
45.0% |
11.8% |
7.08 (4.34, 11.55) p < 0.0001 |
|
CA* wk 9-52< |
20.1% |
3.3% |
9.00 (3.97, 20.41) p < 0.0001 |
*CA : Continuous abstinence rate
Step-by-step approach to smoking cessation
In a 52-week, double-blind, placebo-controlled clinical study, varenicline was evaluated in 1,510 patients who were unable or unwilling to quit within four weeks but were willing to taper their smoking over the first 12 weeks of withdrawal. Patients were randomly assigned to the varenicline group (1 mg twice daily) (n = 760) or placebo (n = 750) for a 24-week treatment period and were followed up to 52 weeks. By the end of the first four weeks of treatment, patients were instructed to reduce their smoking by at least 50%, with a further 50% reduction from the fourth week of treatment to the eighth week, reaching the goal of complete abstinence at 12 weeks. After the initial 12-week reduction phase, patients continued treatment for 12 weeks. Patients treated with this product had significantly higher rates of sustained abstinence compared to placebo. The primary outcomes are summarized in the following table.
Continued withdrawal rates in patients treated with varenicline and placebo
|
This product n=760 |
Placebo N=750 |
ratio (95% confidence interval) p-value |
|
|
CA* wk 15-24 |
32.1% |
6.9% |
8.74 (6.09, 12.53) p<0.0001 |
|
CA* wk 21-52 |
27.0% |
9.9% |
4.02 (2.94 , 5.50) p<0.0001 |
*CA : Continuous abstinence rate
The safety of this product in this study was consistent with premarketing studies.
Patients with co-morbid cardiovascular disease
Varenicline was evaluated in a randomized, double-blind, placebo-controlled clinical study in patients with stable cardiovascular disease (in addition to hypertension) who had been diagnosed with cardiovascular disease 2 months earlier. Patients were randomly assigned to either the varenicline group (1 mg twice daily) (n=353) or the placebo group (n=350) for a 12-week treatment period followed by a 40-week treatment-free follow-up period. 4-week sustained abstinence rates (CQR) were 47.3% in the varenicline group and 14.3% in the placebo group; 9-52-week sustained abstinence rates (CA) were 19.8% in the varenicline group and 7.8% in the placebo group. 19.8% and 7.4% in the placebo group, respectively.
Death and serious cardiovascular events were adjudicated by an independent blinded committee. Adjudicated events with a frequency of ≥1% in each group during treatment (or within 30 days of treatment) were nonfatal myocardial infarction (1.1% in the varenicline group and 0.3% in the placebo group) and hospitalization for angina (0.6% in the varenicline group and 1.1% in the placebo group). During the treatment-free follow-up period lasting up to 52 weeks, adjudicated events included the need for coronary revascularization (2.0 % in the varenicline group, 0.6 % in the placebo group), hospitalization for angina (1.7 % in the varenicline group, 1.1 % in the placebo group), newly diagnosed peripheral vascular disease (PVD) or hospitalization for peripheral vascular disease (1.1 % in the varenicline group, 0.6 % in the placebo group) . Some patients requiring coronary revascularization received management of non-fatal myocardial infarction and hospitalization for angina pectoris. Cardiovascular death occurred in 0.3 % of patients in the varenicline group compared with 0.6 % in the placebo group over 52 weeks of the study.
Patients with combined mild to moderate chronic obstructive pulmonary disease (COPD)
The efficacy and safety of varenicline tartrate (1 mg twice daily) has been demonstrated in a randomized, double-blind, placebo-controlled clinical study in patients with mild to moderate chronic obstructive pulmonary disease. Patients received 12 weeks of treatment during the 52-week study period, followed by a 40-week treatment-free follow-up period. The study’s primary study endpoint was a 4-week sustained abstinence rate (4W CQR) from week 9 to week 12 as confirmed by CO testing, with another key secondary study endpoint of sustained abstinence (CA) from week 9 to week 52. The safety profile of varenicline tartrate (including pulmonary safety) is consistent with that of established clinical studies conducted in normal populations. results for 4-week sustained abstinence rate (week 9 to week 12) and sustained withdrawal (week 9 to week 52) are shown in the following table.
|
4W CQR |
CA (Weeks 9-52) |
|
|
Varenicline tartrate, (n = 248) |
42.3% |
18.5% |
|
Placebo, (n = 251) |
8.8% |
5.6% |
|
Ratio (varenicline tartrate and placebo) |
8.40 p<0.0001 |
4.04 p<0.0001 |
Study conducted in subjects with a history of major depression
The efficacy of varenicline was confirmed in a placebo-controlled randomized trial that included 525 subjects who had suffered from major depression in the past 2 years or who were currently on stable depression treatment. Quit rates in this group were similar to those reported for the general population. At Week 912, the sustained abstinence rate was 35.9% in the varenicline treatment group compared to 15.6% in the placebo group (ratio 3.35 (95% CI 2.165.21)). At Week 952, sustained abstinence rates were 20.3% and 10.4% in the two groups, respectively (Ratios 2.36 (95% CI 1.403.98)). The most common (≥10%) adverse events in subjects in the varenicline group were nausea (27.0% compared with 10.4% in the placebo group), headache (16.8% compared with 11.2%), abnormal dreams (11.3% compared with 8.2%), insomnia (10.9% compared with 4.8%), and irritability (10.9% compared with 8.2%). Psychosis scale results showed no difference between the varenicline and placebo groups, and neither group experienced an overall worsening of depression or other psychotic symptoms during the study period.
Study of patients with comorbid stable schizophrenia or affective schizotypal disorder
A double-blind clinical study evaluating the safety and tolerability of varenicline was conducted in 128 patients with stable schizophrenia or affective schizoaffective disorder who were treated with valium medication. Patients were randomly assigned in a 2:1 ratio to either the varenicline group (1 mg twice daily) or the placebo group for a 12-week treatment period, followed by a 12-week treatment-free follow-up period. The most common adverse events for patients in the varenicline group were nausea (23.8% , 14.0% in the placebo group), headache (10.7% , 18.6% in the placebo group), and vomiting (10.7% , 9.3% in the placebo group). Of the reported neurological adverse events, the only event reported in both groups at ≥5% was insomnia, with a higher rate in the varenicline group than in the placebo group (9.5% in the varenicline group and 4.7% in the placebo group).
Overall, there was no worsening of schizophrenia and no overall change in external pyramidal signs in either group as measured by the psychiatric scale. Compared with the placebo group, a higher proportion of patients in the varenicline group reported suicidal ideation or behavior before enrollment (medical history) and after the end of pharmacotherapy (on days 33 to 85 after the last dose). The incidence of suicide-related events was similar in patients in the varenicline and placebo groups during the drug treatment phase (11% in the varenicline group and 9.3% in the placebo group). In the varenicline group, the proportion of patients who experienced a suicide-related event during the drug treatment and the treatment-free period after treatment did not change; in the placebo group, the proportion of patients who experienced a suicide-related event appeared to decrease during the treatment-free period after treatment. Although there were no suicides, one patient treated in the varenicline group attempted suicide, and the patient’s medical history included several similar episodes. Data from this single clinical study of smoking cessation are limited and insufficient to confirm safety in patients with schizophrenia or affective schizotypal disorder disorders.
A psychoneurological safety study in subjects with and without a history of mental illness
Varenicline was evaluated in a randomized, double-blind, active and placebo-controlled study in subjects with a history of psychiatric disorders (psychiatric group, N=4074) and subjects without a history of psychiatric disorders (non-psychiatric group, N=3984). Subjects were between 1875 years of age, smoked 10 or more cigarettes per day, and were randomly assigned in a 1:1:1:1 ratio to the varenicline 1 mg BID group, the bupropion SR 150 mg BID group, and the nicotine replacement therapy patch (NRT) 21 mg/day group or placebo group in a dose-decreasing fashion for 12 weeks, followed by 12 weeks of post-treatment follow-up.
The primary safety endpoint was the following composite psychoneurological (NPS) adverse events: severe anxiety, depression, sensory abnormalities, or hostile events, and/or moderate or severe agitation, aggression, delusions, hallucinations, homicidal ideation, mania, panic, paranoia, psychosis, suicidal ideation, suicidal behavior, or completed suicidal events.
The table below shows the rates of the primary endpoint of composite NPS adverse events by treatment group and the risk difference (RD) compared with the placebo group in the non-psychiatric group (95% CI).
In addition, this table shows a subset of serious composite NPS adverse event endpoints:
|
Non-psychiatric group |
||||
|
Vaniklang |
Bupropion |
NRT |
Placebo |
|
|
Number of patients treated |
990 |
989 |
1006 |
|
|
Composite NPS AE primary endpoints. n (%) |
13 (1.3) |
22 (2.2) |
25 (2.5) |
24 (2.4) |
|
RD compared to placebo group (95% CI) |
-1.28 (-2.40, -0.15) |
-0.08 (-1.37, 1.21) |
-0.21 (-1.54,1.12) |
|
|
Severe Composite NPS AE n (%) |
1 (0.1) |
4 (0.4) |
3 (0.3) |
5 ( 0.5) |
AE: adverse event; NRT = nicotine replacement therapy patch
Event rates for the composite endpoint were low in all treatment groups and were similar or lower in each active treatment group compared to the placebo group. The use of varenicline, bupropion, and NRT in the nonpsychiatric group did not cause a significantly higher risk of NPS adverse events in the composite primary endpoint compared with the placebo group (95% CI below or including zero).
The percentages of subjects experiencing suicidal ideation and/or behavior based on the Columbia Suicide Severity Scale (C-SSRS) during treatment and during non-treatment follow-up were similar in the varenicline and placebo groups, as shown in the table below.
Non-psychiatric group N=3984 Varenicline
N=990
n (%) Bupropion
N=989
n (%)NRT
N=1006
n (%)Placebo
N=999
n (%) Number of patients assessed during treatment 988983996995 Suicidal behavior and/or ideation7 (0.7)4 (0.4)3 (0.3)7 (0.7) Suicidal behavior 001 (0.1)1 (0.1) Suicidal ideation7 (0.7)4 (0.4)3 (0.3)6 (0.6) Number of patients assessed during follow-up 807816800805 Suicidal behavior and/or /or ideation3 (0.4)2 (0.2)3 (0.4)4 (0.5)Suicidal behavior01 (0.1)00 Suicidal ideation3 (0.4)2 (0.2)3 (0.4)4 (0.5)NRT = nicotine replacement therapy patch
There was one case of completed suicide that occurred while subjects in the non-psychiatric illness group who were treated with placebo were receiving treatment.
The table below shows the rate of the primary endpoint of composite NPS adverse events by treatment group and the risk difference (RD) (95% CI) compared to the placebo group in the psychiatric illness group. Individual events for this endpoint are also shown.
In addition, this table shows a subset of the severe composite NPS AE endpoints.
Mental illness group N=4074 Number of patients treated with varenicline bupropion NRT placebo 1026101710161015 Composite NPS AE primary endpoint, n (%)67 (6.5)
68 (6.7)
53 (5.2)
50 (4.9)
RD compared to placebo group (95% CI) 1.59
(-0.42, 3.59) 1.78
(-0.24, 3.81)0.37
(-1.53, 2.26) NPS AE primary endpoint events n (%).
Anxietya
Depressiona
Sensory abnormalitya
Hostilitya
Agitationb
Aggressivenessb
Delusionsb
Hallucinationsb
Homicidal ideationb
Maniab
Panicb
Paranoiab
Psychosisb
Suicidal behaviorb
Suicidal ideationb
Completed suicideb
5 (0.5)
6 (0.6)
0
0
25 (2.4)
14 (1.4)
1 (0.1)
5 (0.5)
0
7 (0.7)
7 (0.7)
1 (0.1)
4 (0.4)
1 (0.1)
5 (0.5)
0
4 (0.4)
4 (0.4)
1 (0.1)
0
29 (2.9)
9 (0.9)
1 (0.1)
4 (0.4)
0
9 (0.9)
16 (1.6)
0
2 (0.2)
1 (0.1)
2 (0.2)
0
6 (0.6)
7 (0.7)
0
0
21 (2.1)
7 (0.7)
1 (0.1)
2 (0.2)
0
3 (0.3)
13 (1.3)
0
3 (0.3)
0
3 (0.3)
0
2 (0.2)
6 (0.6)
0
0
22 (2.2)
8 (0.9)
0
2 (0.2)
0
6 (0.6)
7 (0.7)
2 (0.2)
1 (0.1)
1 (0.1)
2 (0.2)
0 severe composite NPS AE n (%)14 (1.4)14 (1.4)14 (1.4)14 (1.4)13 (1.3) AE: adverse event; class a = severe adverse event; class b = moderate and severe adverse event; NRT = nicotine replacement therapy patch
Patients in the psychiatric group reported more events in each treatment group compared to the non-psychiatric group, and the event rates were higher in each active treatment group compared to the placebo group for the composite endpoint. However, the use of varenicline, bupropion, and NRT in the mental illness group did not cause a significantly higher risk of NPS adverse events in the composite primary endpoint compared with the placebo group (95% CI includes zero).
The percentages of subjects experiencing suicidal ideation and/or behavior based on the Columbia Suicide Severity Scale (C-SSRS) during treatment and non-treatment follow-up were similar in the varenicline and placebo groups in the mental illness group, as shown in the following table.
Mental illness group N=4074 varenicline
N=1026
n (%) Bupropion
N=1017
n (%) NRT
N=1016
n (%)Placebo
N=1015
n (%) Number of patients assessed during treatment 1017101210061006 Suicidal behavior and/or ideation27 ( 2.7)15 ( 1.5)20 (2.0)25 ( 2.5) Suicidal behavior01 (0.1)02 (0.2) Suicidal ideation27 ( 2.7)15 ( 1.5)20 (2.0)25 ( 2.5) Number of patients assessed during follow-up 833836824791 Suicidal behavior and/or ideation14 (1.7)4 (0.5)9 (1.1)11 (1.4)Suicidal behavior1 (0.1)01 (0.1)1 (0.1)Suicidal ideation14 (1.7)4 (0.5)9 (1.1)11 (1.4)NRT = nicotine replacement therapy patch
No suicide completions were reported in the psychiatric group.
The most commonly reported adverse events in subjects treated with varenicline in this study were similar to those observed in premarketing studies.
In both groups, subjects treated with varenicline showed statistically superior withdrawal rates at week 9 to week 12 and week 9 to week 24 as confirmed by carbon monoxide testing compared to subjects treated with bupropion, NRT patch and placebo (as shown in the table below).
The primary efficacy results are summarized in the following table.
Non-psychiatric group Psychiatric group CA 9-12 n/N (%) Varenicline 382/1005 (38.0%) 301/1032 (29.2%) Bupropion 261/1001 (26.1%) 199/1033 (19.3%) NRT 267/1013 (26.4%) 209/1025 (20.4%) Placebo 138 /1009 (13.7%)117/1026 (11.4%)Treatment comparison: ratio (95% CI), P value Varenicline versus placebo4.00 (3.20, 5.00), P<0.00013.24 (2.56, 4.11) , P<0.0001 Varenicline versus placebo2.26 (1.80, 2.85) , P<0.00011.87 (1.46, 2.39) , P<0.0001 NRT versus placebo 2.30 (1.83, 2.90) , P<0.00012.00 (1.56, 2.55) , P<0.0001 Varenicline versus bupropion 1.77 (1.46, 2.14) , P< 0.00011.74 (1.41, 2.14) , P<0.0001 Varenicline with NRT 1.74 (1.43, 2.10) , P<0.00011.62 (1.32, 1.99) , P<0.0001 CA 9-24 n/N (%) Varenicline 256/1005 (25.5%) 189/ 1032 (18.3%) Bupropion 188/1001 (18.8%) 142/1033 (13.7%) NRT 187/1013 (18.5%) 133/1025 (13.0%) Placebo 106/1009 (10.5%) 85/1026 (8.3%) Treatment comparison: ratio (95% CI), P value Varenicline Lan vs placebo 2.99 (2.33, 3.83), P<0.00012.50 (1.90, 3.29) , P<0.0001 Bupropion vs placebo 2.00 (1.54, 2.59), P<0.00011.77 (1.33, 2.36) , P<0.0001 NRT vs placebo 1.96 (1.51, 2.54), P<0.00011.65 (1.24, 2.20), P=0.0007 Varenicline vs bupropion 1.49 (1.20, 1.85) P=0.00031.41 (1.11, 1.79), P=0.0047 Varenicline vs NRT 1.52 (1.23, 1.89) , P=0.00011.51 (1.19, 1.93), P=0.0008 CA = continuous withdrawal rate; CI = confidence interval; NRT = nicotine replacement therapy patch
Psychoneurosafety meta-analysis and observational studies.
There was no evidence from analysis of clinical trial data that varenicline increased the risk of serious psychoneurological events compared with placebo. In addition, independent observational studies did not support an increased risk of serious psychoneurological events in patients using varenicline compared to those using nicotine replacement therapy (NRT) or bupropion.
Clinical studies in Asians
In a clinical trial involving a total of 15 centers in China, Singapore, and Thailand, a randomized, double-blind, placebo-controlled trial design was used to compare the effectiveness and safety of varenicline versus placebo for smoking cessation. The study period consisted of 24 weeks, including a 12-week treatment phase and a 12-week treatment-free follow-up phase. Approximately 330 subjects were randomized in a 1:1 ratio to receive either varenicline or placebo (1 mg twice daily for 11 weeks after 1 week of dose escalation). At the week 12 visit, dosing was discontinued and the treatment-free follow-up phase was entered until week 24.
Primary endpoint of the study: 4-week sustained abstinence rate (CQR) at weeks 9 to 12, inclusive, as confirmed by CO testing. Two key secondary efficacy endpoints: sustained abstinence (CA) from week 9 to week 24; long-term quit rate (LTQR) from week 9 to week 24. Other secondary efficacy endpoints: 7-day time-point abstinence at weeks 12 and 24, and 4-week time-point abstinence at week 24.
Efficacy outcomes: The primary efficacy endpoint of 4-week sustained abstinence rate confirmed by CO measurement was significantly higher in the varenicline treatment group (50.3%) than in the placebo group (31.6%) (p=0.0003). Key secondary efficacy measures of sustained abstinence (CA) at weeks 9 to 24 and long-term quit rate (LTQR) at week 24, as well as other secondary efficacy measures, were statistically significant between the varenicline and placebo groups. See the following table.
Endpoint varenicline
N=165n (%) Placebo
N=168n (%) Dominance ratio (95% CI) p-value 4-week CQR at weeks 9-12 83 (50.3)53 (31.6)2.31 (1.45, 3.67)0.0003 CA at weeks 9-24 63 (38.2)42 (25.0)1.92 (1.18, 3.13)0.0080 LTQR at weeks 24 73 ( 44.2)45 (26.8)2.29 (1.42, 3.71)0.0006Week 12 7-day time-point abstinence rate104 (63.0)75 (44.6)2.2 (1.41, 3.54)0.0005Week 24 7-day time-point abstinence rate88 (53.3)70 (41.7)1.69 (1.06, 2.68)0.0260 4-week time-point quit rate at week 2487 (52.7)68 (40.5)1.73 (1.09, 2.75)0.0196 Note: 7-day time-point quit rate is defined as the percentage of patients who remained abstinent during the 1-week period prior to the visit.
The 4-week time point quit rate is defined as the proportion of patients who have maintained abstinence within 4 weeks prior to the visit.
Pharmacology and Toxicology
Pharmacological effects
Varenicline is a selective partial agonist of the α4β2 subtype of nicotinic acetylcholine receptors, with high affinity for this receptor in nerves. Varenicline binds to the α4β2 receptor to produce agonistic effects, while blocking the binding of nicotine to this receptor, which is the mechanism by which varenicline exerts its smoking cessation effects.
In vitro electrophysiological studies and in vivo neurochemical studies have shown that varenicline binds to the neural α4β2 nicotinic acetylcholine receptor and stimulates receptor-mediated activity, but this effect is significantly weaker than that of nicotine. Varenicline blocked the activation of α4β2 receptors by nicotine, thereby activating the limbic dopamine system in the midbrain, which is the underlying neural mechanism for the reinforcing-rewarding effects of smoking. Varenicline is highly selective for the α4β2 receptor and binds strongly to this receptor subtype than to other common nicotinic receptors (α3β4 >500-fold, α7>3500-fold, α1βγδ >20,000-fold), non-nicotinic receptors and transporter proteins (>2000-fold). In addition, varenicline has a moderate affinity for 5-HT3 receptors (Ki=350nM).
Toxicological studies
Genotoxicity
The results of varenicline Ames test, mammalian CHO/HGPRT test, human lymphocyte chromosome aberration test, and rat micronucleus test were all negative.
Reproductive toxicity
In fertility and early embryonic development assays, no fertility impairment was observed in male and female rats given varenicline succinate orally at doses up to 15 mg/kg/day (67 and 36 times the maximum recommended daily human exposure of 1 mg BID in male and female rats, respectively, based on AUC). In an embryo-fetal developmental toxicity assay, oral administration of varenicline succinate at doses up to 15 and 30 mg/kg/day (36 and 50 times the maximum recommended human daily exposure of 1 mg BID, respectively, based on AUC) to pregnant rats and rabbits during the organogenesis phase did not show teratogenic effects. In pregnant rabbits, oral administration of varenicline succinate at a dose of 30 mg/kg/day resulted in a reduction in fetal weight; this was not observed at a dose of 10 mg/kg/day (23 times the maximum recommended human daily exposure of 1 mg BID according to the AUC).
In a perinatal toxicity test, oral administration of varenicline succinate at doses up to 15 mg/kg/day from organogenesis to lactation in pregnant rats was associated with reduced offspring fertility and increased auditory startle response. No changes were observed at a dose of 3 mg/kg/day (9 times the maximum recommended human daily exposure of 1 mg BID based on AUC).
Carcinogenicity
No increase in tumor incidence was observed in CD-1 mice given varenicline orally at doses up to 20 mg/kg/day (approximately 47 times the maximum recommended daily human exposure based on AUC) for 2 years.
SD rats were given varenicline orally at doses of 1, 5, and 15 mg/kg/day for 2 years. In male rats (n = 65/sex/dose group), an increased incidence of hibernoma was seen in the medium and high dose groups (1 case at the medium dose of 5 mg/kg/day based on an AUC approximately 23 times the maximum recommended daily exposure in humans, and 2 cases at the high dose of 15 mg/kg/day based on an AUC approximately 67 times the maximum recommended daily exposure in humans). The clinical relevance of this finding to humans has not been established. No increase in tumor incidence was observed in female rats.
[Pharmacokinetics].
Absorption.
Varenicline generally reaches peak plasma concentration 3 to 4 hours after oral administration. After multiple oral doses in healthy volunteers, the blood concentration can reach steady state within 4 days. Oral administration is completely absorbed with high systemic bioavailability.
Food effects.
The oral bioavailability of varenicline is not affected by food or time of administration.
Distribution.
Varenicline is distributed in a variety of tissues, including brain tissue. The steady-state apparent volume of distribution averaged 415 liters (%CV=50). Varenicline plasma protein binding was low (≤20%) and was independent of age and renal function. In rodents, varenicline can pass through the placenta and be secreted in milk.
Biotransformation.
Varenicline is poorly metabolized, with 92% excreted in the urine as the prodrug and less than 10% as metabolites. Small amounts of metabolites in the urine include varenicline – N-carbamoyl glucosinolate and hydroxyvarenicline. Ninety-one percent of the substances associated with varenicline in the body circulation were prodrugs. Small amounts of metabolites in the body circulation included varenicline – N-carbamoyl glucosinolate and N-transglucosyl varenicline.
In vitro studies showed that varenicline did not inhibit cytochrome P450 enzymes (IC50> 6,400 ng/ml). P450 enzymes tested by inhibition assays included 1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1 and 3A4/5. Studies also showed that varenicline did not induce the activity of cytochrome P450 enzymes 1A2 and 3A4 in human isolated hepatocytes. Therefore, varenicline is unlikely to alter the pharmacokinetic parameters of a compound that is primarily metabolized by cytochrome P450 enzymes.
Excretion.
The clearance half-life of varenicline is approximately 24 hours, and its renal excretion is mainly via glomerular filtration and active secretion by the renal tubules with the aid of the organic cation transport protein OCT2.
Linearity/nonlinearity.
Varenicline has linear kinetics when administered as a single dose (0.1-3 mg) or as repeated doses (1-3 mg/day).
Pharmacokinetics in special populations.
The pharmacokinetic parameters of varenicline do not vary in a clinically significant manner by age, race, gender, smoking status or coadministration as demonstrated by specific pharmacokinetic studies and population pharmacokinetic analyses.
Patients with hepatic impairment.
Because varenicline is largely not metabolized by the liver, its pharmacokinetic parameters are not affected when administered to patients with hepatic impairment (see [DOSAGE AND ADMINISTRATION]).
Patients with renal impairment.
In subjects with mild renal impairment (estimated creatinine clearance > 50 ml/min and ≤80 ml/min), varenicline pharmacokinetic parameters were unchanged. Systemic exposure to varenicline was increased 1.5-fold in patients with moderate renal impairment (estimated creatinine clearance ≥30 ml/min and ≤50 ml/min) compared to subjects with normal renal function (estimated creatinine clearance>80 ml/min). For subjects with severe renal impairment (expected creatinine clearance <30 ml/min), systemic exposure to varenicline was increased 2.1-fold. In subjects with end-stage renal disease (ESRD), varenicline is effectively cleared by hemodialysis (see [DOSAGE AND ADMINISTRATION]).
Geriatric patients.
In elderly patients (65 to 75 years of age) with normal renal function, the pharmacokinetic parameters of varenicline are similar to those of young adult subjects (see [DOSAGE AND ADMINISTRATION]). For elderly patients with reduced renal function, please refer to [DOSAGE AND ADMINISTRATION].
Pediatric Patients.
Adolescents.
Single and multiple dose pharmacokinetic studies have been conducted at ages 12-17 years inclusive, with pharmacokinetic parameters largely proportional to dose over the daily dose range of 0.5 mg to 2 mg. In adolescent patients weighing >55 kg, stable systemic exposure to varenicline was evaluated using the area under the drug-time curve (AUC0-24), and the results were comparable to those of the adult population at the same dose. At a dose of 0.5 mg twice daily, adolescent patients weighing ≤55 kg had a higher average daily exposure to stable varenicline than adults (approximately 40% higher). The efficacy and safety of varenicline in children under 18 years of age has not been conclusively demonstrated, therefore, there is no recommended dose (see [Dosage and Administration]).
Storage
Seal and store below 25℃.
Packaging
Quit smoking starter pack (aluminum-plastic packaging)
0.5mgx11 tablets and 1mg x14 tablets/box
Cessation maintenance pack (aluminum-plastic packaging)
0.5 mg x28 tablets/box
1 mg x 28 tablets/box
1 mg x56 tablets/box
[Expiration date
36 months
[Executive Standard
Imported drug registration standard JX20130247 and in accordance with the requirements of the Chinese Pharmacopoeia, 2015 edition
[Approval number].
Imported drug registration number: 0.5mgx11 tablets and 1mg x14 tablets/box: H20150041
0.5 mg x 28 tablets/box: H20150039
1 mg x 28 tablets/box, 1 mg x 56 tablets/box: H20150040
[Manufacturer].
Company name: Pfizer Limited
Company Address: Ramsgate Road, Sandwich, Kent CT13 9NJ, United Kingdom
Manufacturer: R-Pharm Germany GmbH
Business address: Heinrich-Mack-Strasse 35, D-89257 Illertissen, Germany
Domestic contact address.
8-13F, Tower B, Minmetals Plaza, 3-7 Chaoyangmen North Street, Dongcheng District, Beijing
Postal code: 100010
Tel: 010-85167000
Product Inquiry Hotline: 400 910 0055