Date of approval:
Olaparib Tablets Instructions
Please read the instructions carefully and use under the guidance of a physician
[Drug Name]
Generic Name: Olaparib Tablets
Trade Name: Lipitor®/LYNPARZA®
English Name: Olaparib Tablets
Hanyu Pinyin: Aolapali Pian
[Ingredients] The active ingredient of this product is Olaparib
Chemical name: 4-(3-{[4-(cyclopropylcarbonyl)piperazin-1-yl]carbonyl}4-fluorophenyl)methyl]phthalazin-1(2H)-one
Chemical structure formula.
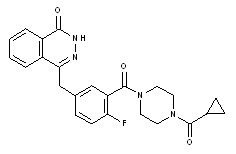
Molecular Formula: C24 H23 FN4 O3
Molecular weight: 434.46
[Properties]This product is a film-coated tablet
150 mg: Green to green/gray, oval, biconvex tablet with “OP150” engraved on one side and blank on the other side.
100 mg: yellow to dark yellow, oval, biconvex tablets with “OP100” engraved on one side and blank on the other side.
[Indications]
.
This product is indicated for the maintenance treatment of adult patients with platinum-sensitive recurrent epithelial ovarian, fallopian tube, or primary peritoneal cancer after achieving complete or partial remission with platinum-containing chemotherapy.
[Specifications](1) 150 mg; (2) 100 mg
This product should be used under the guidance of a physician experienced in the use of antineoplastic drugs.
Recommended Dose
This product is available in 150 mg and 100 mg sizes.
The recommended dose is 300 mg (2 150 mg tablets) twice daily, corresponding to a total daily dose of 600 mg. 100 mg tablets are intended for use in dose reductions.
Patients should begin treatment with this product within 8 weeks of the end of platinum-containing chemotherapy and continue treatment until disease progression or unacceptable toxicities occur.
Method of Administration
Oral administration. This product should be swallowed whole and the tablet should not be chewed, crushed, dissolved, or broken. This product may be taken with a meal or on an empty stomach.
missed dose
If a patient misses a dose of medication, the next dose should be taken at the scheduled time as normal.
Dose Adjustment
For adverse events
To manage adverse events, such as nausea, vomiting, diarrhea, or anemia, consider interrupting therapy or reducing the dose.
If a dose reduction is required, the recommended dose is reduced to 250 mg (1 150 mg tablet and 1 100 mg tablet) taken twice daily (equivalent to a total daily dose of 500 mg).
If further dose reduction is required, the recommended dose is reduced to 200 mg (2 100 mg tablets) taken twice daily (equivalent to a total daily dose of 400 mg).
Combination of cytochromeP450(CYP) em>)3AInhibitors
Combination of strong or moderately potent CYP3A inhibitors is not recommended when using this product, and other alternative medications should be considered. If a potent CYP3A inhibitor must be combined, it is recommended that the dose of this product be reduced to 100 mg (1 100 mg tablet) twice daily (equivalent to a total daily dose of 200 mg). If a combination of intermediate-acting CYP3A inhibitors is necessary, a dose reduction to 150 mg (1 150 mg tablet) twice daily (equivalent to a total daily dose of 300 mg) is recommended (see [Precautions] and [Drug Interactions]).
Medication for Special Populations
Renal Impairment:
In patients with mild renal impairment (creatinine clearance 51-80 mL/min), this product can be used without dose adjustment; in patients with moderate renal impairment (creatinine clearance 31-50 mL/min), the recommended dose of this product is 200 mg (2 100 mg tablets) twice daily (equivalent to a total daily dose of 400 mg); this product is not available for use in patients with severe renal No data are available on the safety and efficacy of this product in patients with severe renal impairment or end-stage renal disease (creatinine clearance ≤ 30 mL/min), and its use is not recommended (see [Pharmacokinetics]).
Hepatic Impairment:
This product may be used in patients with mild hepatic impairment (Child-Pugh classification A) without dose adjustment (see [Pharmacokinetics]). There are no safety and efficacy data for this product in patients with moderate or severe hepatic impairment, and its use is not recommended (see [Pharmacokinetics]).
Children or Adolescents:
The safety and efficacy of this product in children and adolescents have not been established, and it is not recommended for pediatric patients.
Elderly (>65 years):
No starting dose adjustment is required in elderly patients. Limited clinical data are available for patients 75 years of age and older.
[Adverse Reactions]
Because clinical trials are conducted under a variety of different conditions, the incidence of adverse reactions observed in clinical trials for one drug is not directly comparable to the incidence of adverse reactions observed in clinical trials for another drug and does not reflect the incidence observed in clinical practice.
Maintenance Therapy for Recurrent Ovarian Cancer
The following adverse reactions were reported from clinical trials conducted in 558 patients with ovarian cancer (331 received olaparib and 227 received placebo).
SOLO-2
In the SOLO-2 study, the safety of this product was evaluated for maintenance therapy in patients with platinum-sensitive germline breast cancer susceptibility mutations (gBRCAm) ovarian cancer. The study was a placebo-controlled, double-blind study in which 294 patients received either 300 mg of this product (2×150 mg tablets) twice daily (n=195) or placebo tablets twice daily (n=99) until disease progression or intolerable toxicity developed. The median duration of study treatment was 19.4 months for patients receiving this product and 5.6 months for patients receiving placebo. Treatment interruptions due to adverse reactions of any grade occurred in 45% of patients treated with this product compared to 18% in the placebo group, and dose reductions due to adverse reactions occurred in 27% of patients compared to 3% in the placebo group. The most common adverse reactions leading to treatment interruption or dose reduction included anemia (22%), neutropenia (9%), and fatigue/weakness (8%). Treatment with this product resulted in discontinuation in 11% of patients compared to 2% in the placebo group.
Table 1 summarizes the adverse reactions that occurred in at least 20% of patients treated with this product in SOLO-2. Table 2 lists the abnormal laboratory tests that occurred in at least 25% of patients treated with SOLO-2 receiving this product.
Table1 Adverse Reactions in SOLO-2a (≥20%patients treated with olaparib tablets)
Adverse reactions
Olaparib tablets
n=195
Placebo
n=99
1-4Grades
%
Grades 3-4
%
Grades 1-4
%
Grades 3-4
%
Diseases of the blood and lymphatic system
Anemiab
44
20
9
2
Gastrointestinal Disorders
Nausea
76
3
33
0
Vomiting
37
3
19
1
Diarrhea
33
2
22
0
Oral mucositisc
20
1
16
0
Infectious and Infectious Diseases
Nasopharyngitis/upper respiratory tract infection/sinusitis/rhinitis/influenza
36
0
29
0
Systemic disease and various reactions at the site of administration
Fatigue (including weakness)
66
4
39
2
Metabolic and nutritional disorders
Loss of appetite
22
0
11
0
Various musculoskeletal and connective tissue disorders
Arthralgia/Myalgia
30
0
28
0
All types of neurological disorders
Taste Disorders
27
0
7
0
Headache
26
1
14
0
a Graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events (CTCAE) version 4.0
b represents categorical terms that include anemia, decreased erythrocyte pressure volume, decreased hemoglobin, iron deficiency, increased mean cell volume, and decreased red blood cell count
c stands for categorical terms including oral abscess, orofacial ulcer, gingival abscess, gum disease, gum pain, gingivitis, oral ulcer, mucosal infection, mucosal inflammation, oral candidiasis, oral discomfort, oral herpes, oral infection, oral mucosal erythema, oral pain, oropharyngeal discomfort, and oropharyngeal pain
In addition, adverse reactions that occurred in 20% of patients treated with this product in SOLO-2<were neutropenia, rash, cough, dyspepsia, leukopenia, hypomagnesemia, dizziness, thrombocytopenia, elevated serum creatinine, lymphopenia, and edema.
Table2 Abnormal laboratory tests reported in ≥25% of patients with SOLO-2
Laboratory indicatorsa
Olaparib Tablets
nb=195
Placebo
nb=99
1-4Grades
%
Grades 3-4
%
Grades 1-4
%
Grades 3-4
%
Average red blood cell volume increasec
89
–
52
–
Lowered hemoglobin
83
17
69
0
Lowered white blood cell count
69
5
48
1
Decreased lymphocyte count
67
11
37
1
Decreased absolute neutrophil count
51
7
34
3
Elevated serum creatinine
44
0
29
0
Lower platelet count
42
2
22
1
a Patients with laboratory test values of CTCAE grade 1 were allowed to be enrolled in the clinical study.
b Represents the number of people in the safety data set, with data in the table based on each laboratory metric for all evaluable patients.
c Represents the proportion of subjects with a mean erythrocyte volume>upper limit of normal (ULN)
Study19
Among patients treated with olaparib capsules, 35% had treatment interruptions due to adverse effects compared with 10% in the placebo group; 26% required dose reduction compared with 4% in the placebo group; and 6% led to discontinuation compared with 2% in the placebo group.
Table 3 summarizes the adverse reactions that occurred in at least 20% of the patients treated with olaparib in Study 19. Table 4 lists the abnormal laboratory tests that occurred in at least 25% of the patients treated with olaparib in Study 19.
Table3 Adverse Reactions in Study19a< strong>(≥
20% of patients treated with olaparib)
Adverse reactions
Olaparib capsules
n=136
Placebo
n=128
1-4Grades
%
Grades 3-4
%
Grades 1-4
%
Grades 3-4
%
Diseases of the blood and lymphatic system
Anemiab
23
7
7
1
Gastrointestinal Disorders
Nausea
71
2
36
0
Vomiting
35
2
14
1
Diarrhea
28
2
25
2
Constipation
22
1
12
0
Systemic disease and various reactions at the site of administration
Fatigue (including weakness)
63
9
46
3
Infectious and Infectious Diseases
Respiratory tract infections
22
2
11
0
Metabolic and nutritional disorders
Loss of appetite
21
0
13
0
All types of neurological disorders
Headaches
21
0
13
1
a According to the National Cancer Institute (NCI) CTCAE 4.0 classification
b Categorical terms representing related terms that reflect the medical concept of adverse reactions.
In addition, among patients treated with this product in Study 19,<20% experienced adverse reactions of dyspepsia, oral mucositis, taste disturbance, dizziness, elevated blood creatinine, neutropenia, thrombocytopenia, leukopenia, lymphocytopenia, dyspnea, fever, and edema.
Table4 Laboratories reported by ≥25% of patients in study19 tests were abnormal
Laboratory indicatorsa
Olaparib capsules
nb=136
Placebo
nb=128
1-4Grades
%
Grades 3-4
%
Grades 1-4
%
Grades 3-4
%
Lowered hemoglobin
82
8
58
1
Increased mean red blood cell volumec
82
–
51
–
Lowered white blood cell count
58
4
37
2
Decreased lymphocyte count
52
10
32
3
Decreased absolute neutrophil count
47
7
40
2
Elevated serum creatinine
45
0
14
0
Lower platelet count
36
4
18
0
a Patients with laboratory test values of CTCAE grade 1 were allowed to be enrolled in the clinical study
b Represents the number of people in the safety data set, with data in the table based on each laboratory metric for all evaluable patients.
c Represents the proportion of subjects with a mean erythrocyte volume> upper limit of normal (ULN)
Description of specific adverse reactions
Hematologic Toxicity
Overall anemia and other hematologic toxicities were low grade (CTCAE grade 1 or 2), however, CTCAE grade 3 and higher events were still reported.
The median time to first episode of anemia was approximately 4 weeks (approximately 7 weeks for CTCAE ≥ grade 3 events). Anemia can be controlled by interruption of therapy and dose reduction (see [Dosage]) and, if appropriate, transfusion. In the SOLO2 study, the incidence of anemia was 43.6% (19.5% for CTCAE ≥3), and the incidence of dose interruptions, dose reductions, and discontinuations due to anemia was 16.9%, 8.2%, and 3.1%, respectively; 17.9% of patients treated with olaparib required one or more transfusions. A dose-effect relationship between olaparib and hemoglobin reduction has been demonstrated. The incidence of change (decline) in hemoglobin relative to baseline CTCAE grade ≥ 2 was 20%, absolute neutrophil values were 15%, platelets were 5%, lymphocytes were 30%, and leukocytes were 20% (all percentages are approximate %) in clinical studies with olaparib tablets.
The incidence of an increase in mean red blood cell volume from low or normal values at baseline to above the upper limit of normal was approximately 55%, returning to normal after discontinuation of treatment without any clinically meaningful effects.
Complete blood count testing at baseline and subsequent monthly monitoring is recommended during the first 12 months of treatment, followed by periodic monitoring for clinically meaningful changes in parameters that occur during treatment. Dosing interruptions or reductions and/or further therapy may be required based on these changes (see [Dosage] and [Precautions]).
Other Laboratory Findings
In clinical studies of this product, the incidence of CTCAE ≥ grade 2 elevation in blood creatinine values compared with baseline was approximately 15%. Data from a double-blind placebo-controlled clinical study showed that the median creatinine value was elevated by 23% compared with baseline and remained at that level during treatment, returning to baseline after discontinuation of treatment with no clinically significant sequelae. 90% of patients had a CTCAE grade 0 level at baseline and an additional 10% had a CTCAE grade 1 level at baseline.
Nausea and vomiting
Nausea usually occurs very early in treatment, with the first nausea occurring within the first month of treatment initiation in most patients. Vomiting usually occurs early in treatment, with the first vomiting occurring within two months of treatment initiation in the majority of patients. Nausea and vomiting are reported intermittently in the vast majority of patients and can be controlled with dose interruptions, dose reductions, and/or antiemetic therapy. Prophylactic antiemetics are not required.
[Contraindicated]
Contraindicated in persons with hypersensitivity to the active drug ingredient or any of the excipient ingredients. Discontinue breastfeeding during treatment and for 1 month after the last dose (see [Use in Pregnant and Lactating Women]).
[Precautions]Hematologic Toxicity
Hematologic toxicity, including clinical diagnosis and/or laboratory findings of mild or moderate (CTCAE grade 1 or 2) anemia, neutropenia, thrombocytopenia, and lymphocytopenia, has been reported in patients treated with this product. Patients should not start treatment with this product until hematologic toxicity from prior antineoplastic therapy has recovered (hemoglobin, platelet and neutrophil levels should recover to ≤ CTCAE grade 1). Whole blood cell testing is recommended at baseline for the first 12 months of treatment, followed by monthly monitoring, and thereafter periodic monitoring for clinically significant changes in parameters that occur during treatment (see [Adverse Reactions]).
If a patient develops severe or transfusion-dependent hematologic toxicity, treatment should be interrupted and relevant hematologic testing should be performed. If clinically abnormal blood markers remain after 4 weeks of interruption of this drug administration, bone marrow analysis and/or blood cytogenetic analysis is recommended.
Myelodysplastic Syndrome/Acute Myeloid Leukemia
In clinical studies, including long-term survival follow-up, the incidence of myelodysplastic syndrome/acute myeloid leukemia (MDS/AML) was <1.5% in patients receiving this product as monotherapy. The majority of events had an outcome of death. In patients who developed MDS/AML, the duration of olaparib treatment ranged from <6 months to >2 years, with limited data on longer exposures. All patients had underlying factors contributing to MDS/AML and had received prior platinum-based chemotherapy. Some patients had also been treated with other DNA-damaging drugs as well as radiotherapy. The vast majority of patients were gBRCA 1/2 mutation carriers. Some of these patients have a previous history of tumor or bone marrow dysplasia. If MDS and/or AML is diagnosed during olaparib tablet therapy, it is recommended that olaparib tablet therapy should be discontinued and the patient should be treated appropriately.
Non-infectious Pneumonia
The incidence of noninfectious pneumonia (including events with an outcome of death) in patients receiving monotherapy with this product in clinical studies<1.0%. The reported clinical presentation of noninfectious pneumonia was variable and was influenced by many pathogenic triggers (lung cancer and/or metastatic lung cancer, underlying lung disease, history of smoking, and/or history of prior chemotherapy and radiation therapy). If a patient develops new or worsening respiratory symptoms such as dyspnea, cough and fever, or abnormal chest imaging findings, interrupt treatment temporarily and begin relevant investigations immediately. If a diagnosis of non-infectious pneumonia is confirmed, treatment should be discontinued and the patient treated appropriately.
Embryonic–Fetal Toxicity
Based on the mechanism of action of this product (poly ADP ribose polymerase inhibition) and animal studies, it can cause fetal harm when administered to pregnant females. Preclinical studies in rats have shown adverse effects of olaparib on embryo-fetal survival. At exposures below the recommended human dose of 300 mg twice daily, severe fetal malformations can be induced.
This product should not be taken during pregnancy. If the patient becomes pregnant while taking the drug, the patient should be informed of the potential fetal hazard of this product. It is recommended that women of childbearing age be advised to use effective contraception during treatment and for 6 months after the last dose.
It is recommended that male patients and their female partners of childbearing potential be advised that they must use effective contraception during treatment and for 3 months after the last dose, and that they cannot donate sperm (see [Medications for Pregnant and Lactating Women]).
Interactions with other drugs
Combining this product with a strong or moderately potent CYP3A inhibitor is not recommended (see [Dosage]). If a combination of a potent or intermediate-acting CYP3A inhibitor is necessary, the dose should be reduced (see [DOSAGE AND ADMINISTRATION]).
Combining this product with a potent or intermediate-acting CYP3A inducer is not recommended. The prescribing physician should be aware that the efficacy of this product may be significantly reduced in patients already receiving this product who require treatment with a potent or intermediate-acting CYP3A inducer (see [Drug Interactions]).
Effect on the ability to drive and operate machinery
Studies of the effects of olaparib on the ability to drive and operate machinery have not been performed. However, weakness, fatigue, and dizziness have been reported during treatment with this product, and patients experiencing these symptoms should drive or operate machinery with caution.
Effect onQTInterval
The effect of olaparib on cardiac repolarization was assessed in 119 patients after a single dose of 300 mg and in 109 patients after multiple doses of 300 mg twice daily. No clinically relevant effect of olaparib on the QT interval was found.
[Pregnancy and lactation
[Use in Pregnant and Lactating Women]
Contraception
Women of childbearing potential should not become pregnant at the start of and during treatment with this product, and a pregnancy test is required prior to the start of treatment. Women of childbearing potential must use effective contraception during treatment and for 6 months after the last dose of this product (see [Precautions]). It cannot be excluded that olaparib may reduce exposure to CYP2C9 substrates through enzyme induction and that the efficacy of hormonal contraceptives may be reduced when combined with olaparib. Therefore, other non-hormonal contraceptive measures should be considered during treatment and pregnancy tests should be performed periodically (see [Drug Interactions]).
Based on genotoxicity and animal reproductive toxicity studies of this product, it is recommended that male patients (whose spouses are women of childbearing age or pregnant women) should use effective contraception during treatment and for 3 months after the last dose of olaparib and should not donate sperm (see [Precautions]).
Pregnancy
Animal studies have shown reproductive toxicity, including severe teratogenic effects and effects on embryo-fetal viability in rat tests where maternal systemic exposure was lower than human exposure at therapeutic doses (see [Pharmacologic Toxicology]). There are no data on the use of olaparib in pregnant women, but based on the mechanism of action of olaparib, olaparib tablets should not be used during treatment and for 6 months after the last dose of this product in women of childbearing potential who are pregnant and not using reliable contraception.
Lactation
Animal studies of olaparib secretion into breast milk have not been conducted. It is not known whether olaparib or its metabolites are secreted into human breast milk, and based on the pharmacological profile of this product, it is recommended that breastfeeding be discontinued during treatment with olaparib tablets and for 1 month after the last dose (see [Contraindications]).
Fertility
Clinical data on fertility are not available. Animal studies have shown no effect of study drug on conception, but adverse effects on embryo-fetal survival (see [Pharmacology and Toxicology]).
[Pediatric Dosage]
The safety and efficacy of this product in children and adolescents have not been established; therefore, this product is not indicated for pediatric use.
[Geriatric Use]
In clinical studies enrolling 687 patients with advanced solid tumors receiving olaparib tablets 300 mg twice daily as monotherapy, 146 (21%) patients were ≥65 years of age, including 29 (4%) patients ≥75 years of age, and no patients were ≥85 years of age. There were no significant differences in safety and efficacy data between younger patients and older patients treated with olaparib.
[Drug Interactions]
Pharmacodynamic Interactions
Clinical studies combining this product with other antitumor agents, including those that damage DNA, have shown enhanced and prolonged myelosuppressive toxicity. The recommended monotherapy dose does not apply to its combination with antineoplastic agents with myelosuppressive properties.
No studies have been performed combining olaparib with vaccines or immunosuppressive agents. Therefore, caution should be exercised when the above drugs are co-administered with olaparib tablets and patients should be monitored closely.
Pharmacokinetic Interactions
In vitro studies have confirmed that olaparib is an inhibitor and inducer of CYP3A and an inducer of CYP2B6. Olaparib is predicted to be a weak CYP3A inhibitor in humans. In vitro studies have also shown that olaparib is an inhibitor of uridine diphosphate glucuronyl transferase (UGT) 1A1, breast cancer resistance protein (BCRP), organic anion transport protein (OATP) 1B1, organic cation transport protein (OCT) 1, OCT2, organic anion transport protein (OAT) 3, multidrug and toxic compound efflux transport protein (MATE) 1 and MATE2K inhibitors. The clinical relevance of these findings is unknown. In in vitro studies, olaparib is a substrate for the efflux transporter P-glycoprotein (P-gp) and inhibits P-gp. The potential for olaparib to induce P-gp has not been evaluated.
Drugs that may elevate olaparib plasma concentrations
Olaparib is primarily metabolized by CYP3A. In patients (n=57), combined use of itraconazole, a potent CYP3A inhibitor, increased olaparib AUC by 170%. The medium-acting CYP3A inhibitor fluconazole was predicted to increase the area under the plasma concentration versus time curve (AUC) of olaparib by 121%.
Avoid the combination of potent CYP3A inhibitors such as itraconazole, telithromycin, clarithromycin, ketoconazole, voriconazole, nefazodone, posaconazole, ritonavir, lopinavir/ritonavir, indinavir, saquinavir, nelfinavir, boceprevir, telaprevir, or intermediate-acting CYPA inhibitors such as amprenavir (amprenavir), aripitant, atazanavir, ciprofloxacin, crizotinib, darunavir/ritonavir, diltiazem, erythromycin, fluconazole, fosamprenavir, imatinib, verapamil. Olaparib dose should be reduced if a combination of potent or moderately potent CYP3A inhibitors is necessary.
Avoid grapefruit, grapefruit juice, lime and lime juice during olaparib treatment because these foods contain CYP3A inhibitors.
Drugs That May Reduce Olaparib Plasma Concentrations
In patients (n=22), combined use of rifampin, a potent CYP3A inducer, reduced olaparib AUC by 87%. A predicted reduction in olaparib AUC of approximately 60% with intermediate-acting CYP3A inducers.
Avoid combining strong CYP3A inducers such as phenytoin, rifampin, carbamazepine, and St. John’s wort) or intermediate-acting CYP3A4 inducers such as bosentan, efavirenz, etravirine, modafinil, and nafcillin. Olaparib efficacy may be reduced if mid-acting CYP3A inducers cannot be avoided.
[Overdose]
Clinical signs of overdose with this product have not been identified. A small number of patients taking olaparib tablets at daily doses up to 900 mg for two days had no unintended adverse reactions reported. There is no specific management for overdose of this product. If an overdose occurs, the physician should provide symptomatic and generally supportive treatment.
[Clinical Trials]
Maintenance Therapy for Recurrent Ovarian Cancer
The efficacy of olaparib was evaluated in two randomized, placebo-controlled, double-blind, multicenter studies in patients with recurrent ovarian cancer who achieved remission after platinum-containing therapy.
SOLO2Study (D0816C00002)
The SOLO2 study is a randomized, double-blind, placebo-controlled phase III trial evaluating the safety and efficacy of olaparib as maintenance therapy in patients with platinum-sensitive recurrent (PSR) ovarian, fallopian tube, or primary peritoneal cancer with gBRCA1/2 mutations. Enrolled 295 patients with high-grade plasma or endometrioid PSR ovarian cancer who experienced remission (complete remission [CR] or partial remission [PR]) after completion of platinum-containing chemotherapy were randomized in a 2:1 ratio (196 in the olaparib group and 99 in the placebo group) to receive olaparib (300 mg [2 x 150 mg tablets] twice daily) or placebo until disease progression or development of intolerable toxicity.
Patients who had received two or more platinum-containing regimens were enrolled in the study until disease recurrence > 6 months after completion of the penultimate platinum-based chemotherapy. Patients must not have received prior treatment with olaparib or other PARP inhibitors. Patients are allowed to have received prior treatment with bevacizumab, but the treatment regimen used prior to randomization must not have included bevacizumab.
At baseline, all patients carried deleterious or suspected deleterious gBRCA mutations, detected locally (n=236) or by central Myriad CLIA testing (n=59) and subsequently confirmed by BRACAnalysis® CDx (n=286). Large fragment rearrangements were detected in the BRCA1/2 gene in 4.7% (14/295) of randomized patients.
Demographic and baseline characteristics were essentially similar between the olaparib and placebo groups. The median age in both groups was 56 years. Ovarian cancer in greater than 80% of patients was primary. The most common histologic type was plasmacytic (> 90%), and 6% of patients had endometrioid carcinoma. In the olaparib group, 55% of patients had received only second-line therapy in the past, while 45% received third-line therapy or more. In the placebo group, 61% of patients had received prior second-line therapy only and 39% had received third-line therapy or more. The majority of patients had an ECOG physical status score of 0 (81 %). 60% had a platinum-free interval of > 12 months and 40% had > 6-12 months. 47% of patients responded to platinum-containing chemotherapy in complete remission and 53% in partial remission. Seventeen percent and 20% of patients in the olaparib and placebo groups, respectively, had received prior bevacizumab therapy.
The primary study endpoint was progression-free survival (PFS), as measured by the investigators using the Remission Evaluation Criteria in Solid Tumors (RECIST) 1.1 assessment. Secondary study endpoints included time to second disease progression or death (PFS2); OS (overall survival), time to treatment discontinuation or death (TDT), time to first subsequent anticancer treatment or death (TFST), time to second subsequent anticancer treatment start or death (TSST); and health-related quality of life (HRQoL ).
The study met its primary study endpoint with a statistically significant improvement in investigator-assessed PFS in the olaparib group over the placebo group with a risk ratio (HR) of 0.30 (95% CI 0.22-0.41; p<0.0001; median value in the olaparib group 19.1 months vs. 5.5 months in the placebo group). The investigator-assessed PFS results were supported by blinded independent central imaging assessments (HR= 0.25; 95% CI 0.18-0.35; p<0.0001; median value 30.2 months in the olaparib group vs. 5.5 months in the placebo group). at 2 years, 43% of patients treated with olaparib remained progression-free compared with those treated with placebo on the other hand, only 15%.
See Table 5 and Figure 1 for a summary of the primary study endpoint results for patients with gBRCA1/2m PSR ovarian cancer in SOLO2.
Table5 Primary gBRCA1/2m PSR Ovarian Cancer Patients in SOLO2 Summary of Study Endpoint Results (Investigator Assessment)
Olaparib300 mg tabletsbd
(n=196)
Placebo
(n=99)
PFS(63% maturity)
Number of events: total number of patients (%)
107:196 (55)
80:99 (81)
Median time (months) (95% CI)
19.1 (16.3-25.7)
5.5 (5.2-5.8)
HR (95% CI)a
0.30 (0.22-0.41)
P-value (bilateral)
p<0.0001
a HR=Risk Ratio. Value<1 supports olaparib. Analysis stratified by remission response to prior platinum-based chemotherapy (CR or PR) and time to disease progression in penultimate platinum-containing chemotherapy (>6-12 months and >12 months), using log-rank test, was performed.
bd twice daily; PFS progression-free survival; CI confidence interval.
Figure 1 SOLO2: gBRCA1/2m PSR Kaplan-Meier curve for PFS in ovarian cancer patients (63%maturity – investigator assessment)
Time (months) since randomization
Placebobd
Olaparib300 mg bd
Number of patients at risk
Placebobd
Olaparib300 mg bd
bd twice daily; PFS progression-free survival
The secondary endpoints TFST and PFS2 showed sustained and statistically significant improvement in the olaparib group compared with the placebo group (Table 6).
Table6 SOLO2 ingBRCA1/2m PSR Patients Key Secondary Study Summary of endpoint results
Olaparib300 mg tabletsbd
(n=196)
Placebo
(n=99)
TFST(58% maturity)
Number of events: total number of patients (%)
92:196 (47)
79:99 (80)
Median time (months) (95% CI)
27.9 (22.6-NR)
7.1 (6.3-8.3)
HR (95% CI) a
0.28 (0.21-0.38)
P-value* (bilateral)
p<0.0001
PFS2(40% maturity)
Number of events: total number of patients (%)
70:196 (36)
49:99 (50)
Median time (months) (95% CI)
NR (24.1-NR)
18.4 (15.4-22.8)
HR (95% CI) a
0.50 (0.34-0.72)
P value (bilateral)
p=0.0002
* Uncontrolled multiplicity
a HR=risk ratio. Value <1 supports olaparib. Analyses were performed stratified by remission response to prior platinum-based chemotherapy (CR or PR) and time to disease progression in the penultimate platinum-containing chemotherapy (>6-12 months and >12 months), using log-rank tests.
bd twice daily; NR for attainment; CI confidence interval; PFS2 randomized to time to second disease progression or death; and TFST randomized to time to start of first subsequent treatment or death.
Among patients enrolled in the trial with measurable lesions (target lesions at baseline), the objective remission rate was 41% in the olaparib tablet group compared with 17% in the placebo group. Complete remission occurred in 15.0% of patients treated with olaparib tablets who had a lesion (target lesion or non-target lesion at baseline) at study entry, compared with 9.1% of patients receiving placebo.
Patient self-reported outcome (PRO) data showed no difference between patients in the olaparib group and the placebo group based on the Functional Assessment of Cancer Therapy-Ovarian Cancer (FACT-O) Trial Outcome Index (TOI) relative to baseline change assessment.
SOLO2Study (D0816C00002)< strong>–
China Cohort
Chinese patients were enrolled in the SOLO2 study as a separate cohort, and a total of 32 Chinese patients were randomized to receive either 300 mg (2 x 150 mg tablets) twice daily (n=22) or placebo tablets twice daily (n=10) until disease progression or intolerable toxicity developed. Chinese subjects were tested and confirmed to have a gBRCA mutation by the Shenzhen UW Clinical Laboratory. The median age of patients treated with this product was 49 years (range: 37 to 65 years) and the median age of patients treated with placebo was 46.5 years (range: 33 to 67 years). The ECOG physical status score was 0 in 73% of patients in the treatment group and 50% of patients in the placebo group. 59% of all patients achieved complete remission after the last platinum-containing chemotherapy prior to randomization, and 56% of patients had a time to disease progression of 6-12 months after completion of the penultimate platinum-containing chemotherapy. Approximately 23% of patients in the olaparib group and 20% of patients in the placebo group had received 3 or more prior lines of platinum-containing therapy. In the SOLO2 China cohort, this product significantly improved patient PFS (as assessed by the investigators) compared with placebo, with an HR of 0.44 (95% CI: 0.17-1.19; p = 0.0776; median value in the olaparib group was 13.8 months vs. 5.5 months in the placebo group). This result was consistent with independent imaging assessments. Data on patient survival are not yet mature (only 19% of patients had an event).
Table7 SOLO2Chinese CohortgBRCA1/2m PSR Patients With Ovarian Cancer Summary of results for the primary study endpoint (investigator-assessed)
Olaparib Tablets 300 mg Tablets bd
(n=22)
Placebo
(n=10)
PFS(65.6% maturation)
Number of events: total number of patients (%)
14:22 (63.6)
7:10 (70.0)
Median time (months) (95% CI)
13.8 (10.1, 16.6)
5.5 (3.2, 8.4)
Risk ratio HR (95% CI)a
0.44 (0.17, 1.19)
P-value (bilateral)a
p=0.0776
a HR=risk ratio, risk ratio vs p-value based on unstratified proportional risk model.
PFS Progression-free survival
Efficacy and safety in Chinese subjects were consistent with those in non-Chinese subjects.
Study19(D0810C00019< em>)
Study 19 is a randomized, double-blind, placebo-controlled phase II trial evaluating the safety and efficacy of olaparib as maintenance therapy in patients with PSR ovarian cancer, including those with tubal or primary peritoneal cancer, after two or more platinum-based regimens. Two hundred and sixty-five patients with PSR-grade plasma ovarian cancer who achieved remission (CR or PR) after completion of platinum-containing chemotherapy were enrolled and randomized 1:1 (136 in the olaparib group and 129 in the placebo group) to receive olaparib capsules 400 mg (8 x 50 mg tablets) twice daily (capsules were not declared available in China) or placebo until disease progression or intolerable toxicity developed. The primary endpoint was PFS as assessed by the investigators using RECIST 1.0 criteria. secondary efficacy endpoints included OS, disease control rate (DCR, i.e., confirmed CR/PR + SD [disease stabilization]), HRQoL, and disease-related symptoms. Exploratory analyses were performed for TFST and TSST.
The study enrolled patients from the completion of penultimate platinum-containing chemotherapy to disease relapse>6 months. Inclusion requirements excluded identified BRCA1/2 mutations (some patients were retrospectively tested for BRCA mutation status). Patients must not have received prior treatment with olaparib or other PARP inhibitors. Patients were allowed to have received prior treatment with bevacizumab, but the treatment regimen used prior to randomization must not have included bevacizumab. No further treatment with olaparib is allowed after disease progression occurs during olaparib treatment.
Patients with BRCA1/2 mutations were identified using local tests or Myriad CLIA comprehensive BRACAnalysis® tests for blood germline testing or tests performed on tumor samples using tests performed by Foundation Medicine. Large fragment rearrangements were detected in the BRCA1/2 gene in 7.4% (10/136) of randomized patients.
Demographic and baseline characteristics were essentially similar between the olaparib and placebo groups. The median age in both groups was 59 years. 86% of patients had primary ovarian cancer. In the olaparib group, 44% of patients had received only second-line therapy in the past, while 56% had received third-line therapy or more. In the placebo group, 49% of patients had received prior second-line therapy only, while 51% had received third-line therapy or more. The majority of patients had an ECOG physical status score of 0 (77%). 60% of patients had a platinum-free interval of > 12 months and 40% had > 6-12 months. 45% of patients had a complete response to platinum-containing chemotherapy and 55% had a partial remission. Six percent and 5% of patients in the olaparib and placebo groups, respectively, had received prior bevacizumab therapy.
The study met the primary study endpoint with a statistically significant improvement in investigator-assessed PFS in the olaparib group compared with the placebo group, with an HR of 0.35 (95% CI 0.25-0.49; p<0.00001; median value of 8.4 months in the olaparib group vs. 4.8 months in the placebo group). At the time of final OS analysis (maturity 79%, data cutoff [DCO] May 09, 2016), the risk ratio for olaparib versus placebo was 0.73 (95% CI 0.55-0.95; p=0.02138 [prespecified significance level not reached< 0.0095]; median value in the olaparib group was 29.8 months and in the placebo group was (27.8 months). In the olaparib-treated group, 23.5% (n=32/136) of patients received treatment for ≥2 years compared with 3.9% (n=5/128) of patients in the placebo group. Despite the limited number of patients, 13.2% (n=18/136) of patients in the olaparib group received treatment for ≥5 years compared with 0.8% (n=1/128) in the placebo group.
A preplanned subgroup analysis found that patients with BRCA1/2 mutations in ovarian cancer (n=136, 51.3%; including 20 patients identified with somatic tumor BRCA1/2 mutations) had the greatest benefit from olaparib monotherapy maintenance treatment. Clinical benefit was also observed in patients with BRCA1/2 wild-type/variants of unknown significance (BRCA1/2 wt/VUS), although to a lesser extent. No multiple testing was available for subgroup analysis.
Please see Table 8 and Figure 2 for a summary of the results of the primary study end points in patients with BRCA1/2 mutations and BRCA1/2 wt/ VUS PSR ovarian cancer in Study 19.
Table8 Study19 inBRCA1/2< strong> mutations andBRCA1/2 wt/VUS PSR Summary of primary study endpoint results in patients with ovarian cancer
All patientsa
BRCA1/2mutations
BRCA1/2 wt/VUS
Olaparib400 mg
Capsulebd
Placebo
Olaparib400 mg
Capsulebd
Placebo
Olaparib400 mg
Capsulebd
Placebo
PFS-DCO 2010Year06Month30< strong>Day
Number of events: total number of patients
(%)
60:136
(44)
94:129
(73)
26:74
(35)
46:62
(74)
32:57
(56)
44:61
(72)
Median time (months) (95% CI)
8.4
(7.4-11.5)
4.8
(4.0-5.5)
11.2
(8.3-NR)
4.3
(3.0-5.4)
7.4
(5.5-10.3)
5.5
(3.7-5.6)
HR (95% CI)b
0.35 (0.25-0.49)
0.18 (0.10-0.31)
0.54 (0.34-0.85)
P value (bilateral)
p<0.00001
p<0.00001
p=0.00745
a All patients include the following subgroups: BRCA1/2-mutation, BRCA1/2 wt/VUS and BRCA1/2 status unknown (11 patients with unknown status are not included in the
table not as a separate subgroup).
b HR=risk ratio. The value <1 supports olaparib. Analysis was performed using a Cox proportional risk model for treatment, ethnic origin, platinum sensitivity
and remission after last platinum-based chemotherapy as effectors.
wt (wild type) wild type; VUS (variants of uncertain significance) variants of uncertain significance; bd twice daily; PFS progression-free survival; DCO data cutoff; CI confidence interval; NR not reached.
Time (months) since randomization
Placebobd
Olaparib400mg bd
Olaparib400mg bd
Placebobd
Number of patients at risk:
bd twice daily; DCO data cutoff; FAS full analysis set; PFS progression-free survival
Results of key secondary study endpoints for patients with BRCA1/2 mutations and BRCA1/2 wt/VUS PSR ovarian cancer in Study 19 are summarized in Table 9, and all patients are summarized in Table 9 and Figure 3.
Table9 Study19 inBRCA1/2 mutations andBRCA1/2 wt/VUS PSR Summary of results of key secondary study endpoints in patients with ovarian cancer
All patientsa
BRCA1/2mutations
BRCA1/2 wt/VUS
Olaparib400 mg
Capsulebd
Placebo
Olaparib400 mg
Capsulebd
Placebo
Olaparib400 mg
Capsulebd
Placebo
OS – DCO 2016Year05Month09< strong>Day
Number of events: total number of patients (%)
98:136
(72)
112:129
(87)
49:74
(66)
50:62
(81)c
45:57
(79)
57:61
(93)
Median time (months)
(95% CI)
29.8
(26.9-35.7)
27.8
(24.9-33.7)
34.9
(29.2-54.6)
30.2
(23.1-40.7)
24.5
(19.8-35.0)
26.6
(23.1-32.5)
HR (95% CI)b
0.73 (0.55-0.95)
0.62 (0.42-0.93)
0.84 (0.57-1.25)
P-value* (Bilateral)
p=0.02138
p=0.02140
p=0.39749
TFST – DCO 2016Year05Month09< strong>day
Number of events: total number of patients (%)
106:136
(78)
124:128
(97)
55:74
(74)
59:62
(95)
47:57
(83)
60:61
(98)
Median time (months)
(95% CI)
13.3
(11.3-15.7)
6.7
(5.7-8.2)
15.6
(11.9-28.2)
6.2
(5.3-9.2)
12.9
(7.8-15.3)
6.9
(5.7-9.3)
HR (95% CI)b
0.39 (0.30-0.52)
0.33 (0.22-0.49)
0.45 (0.30-0.66)
P-value* (Bilateral)
p<0.00001
p<0.00001
p=0.00006
* There was no multiple testing strategy for subgroup analysis or for TFST in all patients.
a All patients included the following subgroups: BRCA1/2 mutations, BRCA1/2 wt/VUS, and BRCA1/2 status unknown (11 patients with unknown status in
table not as a separate subgroup).
b HR=risk ratio. The value <1 supports olaparib. Analysis was performed using a Cox proportional risk model for treatment, race, platinum sensitivity, and
remission after last platinum-based chemotherapy as effectors.
In the c BRCA mutation subgroup, approximately one-quarter (14/62; 22.6%) of patients in the placebo-treated group were subsequently treated with PARP inhibitors.
wt (wild type) wild type; VUS (variants of uncertain significance) variants of uncertain significance; bd twice daily; OS overall survival; DCO data cutoff; CI: confidence interval; TFST time from randomization to first subsequent treatment or death
Figure 3 Study 19: Kaplan-Meier curves for OS in FAS (79%Maturity, DCO 2016 05 09)
Placebo
Olaparib400mg bd
Olaparib400mg bd
Placebo
Number of patients at risk:
Time since randomization (months)
bd twice daily; DCO data cutoff; FAS full analysis set; OS overall survival
Patient self-reported outcome (PRO) data showed no difference between patients in the olaparib group and the placebo group, as measured by the rate of improvement and rate of worsening in the Functional Assessment of Cancer Therapy-Ovarian Cancer Total Score (Total FACT-O) Trial Outcome Index (TOI).
[Pharmacology and Toxicology]
Pharmacological effects
Olaparib is an inhibitor of poly ADP ribose polymerase (PARP, including PARP1, PARP2 and PARP3). PARPases are involved in normal cellular functions such as DNA transcription and DNA repair. The results of the trial showed that olaparib inhibited the proliferation of tumor cell lines in vitro and the growth of human tumor-bearing mouse xenografts in vivo, and was effective as monotherapy or after platinum-based chemotherapy. Olaparib administration produced stronger cytotoxic and tumor suppressive effects when cell lines and mouse transplantation tumor models had BRCA-associated defects in DNA damage homologous recombination repair or non-BRCA-associated defects in DNA damage homologous recombination repair associated with platinum-based chemotherapeutic response. In vitro studies suggest that the cytotoxic effects of olaparib may involve inhibition of PARPase activity and increased PARP-DNA complex formation, leading to DNA damage and cancer cell death.
Toxicological studies
Genotoxicity: Olaparib Ames test results were negative; Chinese hamster ovary (CHO) cell chromosome aberration test and rat bone marrow micronucleus test results were positive, and chromosome breakage effects were seen, consistent with the pharmacological effects of olaparib-induced genomic instability, suggesting that olaparib may be genotoxic in humans.
Reproductive toxicity: In a female rat fertility and early embryonic development toxicity assay, oral administration of olaparib at doses up to 15 mg/kg/day (maternal systemic exposure of approximately 7% of human exposure [AUC0-24h] at the clinically recommended dose) from 14 days prior to mating to day 6 of gestation had no effect on No effects on mating or fertility were seen, but increased post-coital abortions were induced.
In a fertility trial in male rats, oral administration of olaparib at doses up to 40 mg/kg/day (systemic exposure of approximately 5% of the clinically recommended human exposure [AUC0-24h]) for at least 70 days had no effect on mating or fertility.
In an embryo-fetal developmental toxicity assay, orally administered olaparib 0.05 and 0.5 mg/kg/day to pregnant rats during the organogenesis phase. Embryo-fetal toxicity was seen at a dose of 0.5 mg/kg/day (maternal systemic exposure of approximately 0.18% of the clinically recommended human exposure [AUC0-24h]) and included increased post-arrival abortion and fusion or absence of the eye (anophthalmia, microphthalmia), vertebrae/ribs (additional ribs or ossification centers, vertebral arches, ribs, and sternum), skull (occipital external fusion) and severe malformations of the diaphragm (hernia). Other abnormalities or variants include incomplete or absent ossification (vertebrae/sternum, ribs, extremities) and abnormalities of vertebrae/sternum, pelvic girdle, lung, thymus, liver, ureter, and umbilical artery. The incidence of ocular, rib, and ureteral abnormalities described above was low when administered at a dose of 0.05 mg/kg/day.
Carcinogenicity: No relevant studies have been performed.
[Pharmacokinetics]
The dosage forms of olaparib include tablets and capsules (capsules have not been declared for marketing in China). The oral bioavailability of the tablet form is higher than that of the capsule form. Population pharmacokinetic analysis has confirmed that steady-state exposure (AUC) was 77% higher after 2 daily doses of 300 mg tablets than after 2 daily doses of 400 mg capsules. geometric means of olaparib AUC and Cmax after a single dose of 300 mg tablets were 42.0 μg*h/mL (n = 204) and 5.8 μg/mL (n = 204), and the geometric mean of steady-state AUC and Cmax were 49.0 μg*h/mL (n = 227) and 7.7 μg/mL (n = 227), respectively, after 2 daily doses of 300 mg tablets. Olaparib PK was time-dependent, with a 15% reduction in steady-state clearance after multiple dosing.
Absorption
Following oral administration of olaparib, absorption was rapid, with median peak plasma concentrations typically reached 1.5 hours after administration. a steady-state AUC mean accumulation ratio of 1.8 was observed after multiple dosing of 300 mg tablets twice daily.
Olaparib systemic exposure (single-dose AUC) increased approximately proportionally with dose when the dose range was 25 mg to 450 mg, with Cmax increasing slightly less than the proportional dose increase in the same dose range.
Co-administration with a high-fat diet resulted in a rate of absorption of olaparib (tmax delayed by 2.5 hours) but did not significantly alter the extent of olaparib absorption (mean AUC increased by approximately 8%).
Distribution
After a single dose of olaparib 300 mg, the mean (± standard deviation) apparent volume of distribution of olaparib was 158 ± 136 L. Olaparib had an in vitro protein binding rate of approximately 82%.
Metabolism
In in vitro studies, CYP3A4/5 has been shown to be the enzyme primarily responsible for olaparib metabolism.
In female patients, 14C-olaparib accounts for the majority (70%) of the radioactivity circulating in plasma after oral administration of prototype olaparib. 14C-olaparib is extensively metabolized, with the prototype drug accounting for 15% and 6% in urine and feces, respectively. Most metabolism is attributable to oxidative reactions with many of the components produced undergoing subsequent glucosinolate or sulfate binding.
Excretion
After a single dose of olaparib 300 mg, the mean plasma terminal half-life (± standard deviation) was 14.9 ± 8.2 hours, and the apparent plasma clearance was 7.4 ± 3.9 L/h.
After 14C-olaparib single administration, 86% of the given radioactivity was recovered within the 7-day collection period, 44% via urine, and 42% via feces. Most material was excreted as metabolites.
Special Populations
Patient age, sex, weight, or race (including Caucasian, Chinese, and Japanese) were not significant covariates in the population pharmacokinetic analysis.
Hepatic impairmentIn a trial of hepatic impairment, when olaparib was given to patients with mild hepatic impairment (Child-Pugh classification A; n=9), there was a 15% increase in mean AUC and a 13% increase in mean Cmax compared with patients with normal hepatic function (n=13). Mild hepatic impairment had no effect on the protein binding of olaparib, so total plasma exposure represents free drug. Data are not available in patients with moderate or severe hepatic impairment.
Renal Impairment
In a trial of renal impairment, compared with patients with normal renal function (CLcr ≥81 mL/min; n=12), patients with mild renal impairment (as defined by the Cockcroft-Gault equation, CLcr = 51-80 mL/min; n=13) taking olaparib had a 24% increase in mean AUC, a mean Cmaxincreased by 15%, and patients with moderate renal impairment (CLcr = 31-50 mL/min; n=13) had a mean AUC and Cmaxincreased by 44% and 26%, respectively, after taking olaparib. There was no evidence of a correlation between the degree of olaparib plasma protein binding and creatinine clearance. No data were available for patients with severe renal impairment or end-stage renal disease (CLcr ≤30 mL/min).
[Storage].
Store below 30°C.
[Packaging]
Aluminum-aluminum blister pack, 56 tablets per box (7 plates)
Aluminum-aluminum blister pack, 112 tablets per box (14 plates)
[Expiration date]
36 months
[Executive Standard]
Imported drug registration standards.
[Approval Number]
Company name: AbbVie Deutschland GmbH & Co. KG
Production Address: Knollstrasse 67061 Ludwigshafen, Germany
Address of China Liaison Office: No. 2 Huangshan Road, Wuxi New District, Jiangsu Province
Postal code: 214028
Quality complaints: 400 828 1755, 800 828 1755
Product information toll-free: 400 820 8116, 800 820 8116
Fax: 021-38723255
Website: www.astrazeneca.com.cn
[Properties]This product is a film-coated tablet
150 mg: Green to green/gray, oval, biconvex tablet with “OP150” engraved on one side and blank on the other side.
100 mg: yellow to dark yellow, oval, biconvex tablets with “OP100” engraved on one side and blank on the other side.
[Indications]
.
This product is indicated for the maintenance treatment of adult patients with platinum-sensitive recurrent epithelial ovarian, fallopian tube, or primary peritoneal cancer after achieving complete or partial remission with platinum-containing chemotherapy.
[Specifications](1) 150 mg; (2) 100 mg
This product should be used under the guidance of a physician experienced in the use of antineoplastic drugs.
Recommended Dose
This product is available in 150 mg and 100 mg sizes.
The recommended dose is 300 mg (2 150 mg tablets) twice daily, corresponding to a total daily dose of 600 mg. 100 mg tablets are intended for use in dose reductions.
Patients should begin treatment with this product within 8 weeks of the end of platinum-containing chemotherapy and continue treatment until disease progression or unacceptable toxicities occur.
Method of Administration
Oral administration. This product should be swallowed whole and the tablet should not be chewed, crushed, dissolved, or broken. This product may be taken with a meal or on an empty stomach.
missed dose
If a patient misses a dose of medication, the next dose should be taken at the scheduled time as normal.
Dose Adjustment
For adverse events
To manage adverse events, such as nausea, vomiting, diarrhea, or anemia, consider interrupting therapy or reducing the dose.
If a dose reduction is required, the recommended dose is reduced to 250 mg (1 150 mg tablet and 1 100 mg tablet) taken twice daily (equivalent to a total daily dose of 500 mg).
If further dose reduction is required, the recommended dose is reduced to 200 mg (2 100 mg tablets) taken twice daily (equivalent to a total daily dose of 400 mg).
Combination of cytochromeP450(CYP) em>)3AInhibitors
Combination of strong or moderately potent CYP3A inhibitors is not recommended when using this product, and other alternative medications should be considered. If a potent CYP3A inhibitor must be combined, it is recommended that the dose of this product be reduced to 100 mg (1 100 mg tablet) twice daily (equivalent to a total daily dose of 200 mg). If a combination of intermediate-acting CYP3A inhibitors is necessary, a dose reduction to 150 mg (1 150 mg tablet) twice daily (equivalent to a total daily dose of 300 mg) is recommended (see [Precautions] and [Drug Interactions]).
Medication for Special Populations
Renal Impairment:
In patients with mild renal impairment (creatinine clearance 51-80 mL/min), this product can be used without dose adjustment; in patients with moderate renal impairment (creatinine clearance 31-50 mL/min), the recommended dose of this product is 200 mg (2 100 mg tablets) twice daily (equivalent to a total daily dose of 400 mg); this product is not available for use in patients with severe renal No data are available on the safety and efficacy of this product in patients with severe renal impairment or end-stage renal disease (creatinine clearance ≤ 30 mL/min), and its use is not recommended (see [Pharmacokinetics]).
Hepatic Impairment:
This product may be used in patients with mild hepatic impairment (Child-Pugh classification A) without dose adjustment (see [Pharmacokinetics]). There are no safety and efficacy data for this product in patients with moderate or severe hepatic impairment, and its use is not recommended (see [Pharmacokinetics]).
Children or Adolescents:
The safety and efficacy of this product in children and adolescents have not been established, and it is not recommended for pediatric patients.
Elderly (>65 years):
No starting dose adjustment is required in elderly patients. Limited clinical data are available for patients 75 years of age and older.
[Adverse Reactions]
Because clinical trials are conducted under a variety of different conditions, the incidence of adverse reactions observed in clinical trials for one drug is not directly comparable to the incidence of adverse reactions observed in clinical trials for another drug and does not reflect the incidence observed in clinical practice.
Maintenance Therapy for Recurrent Ovarian Cancer
The following adverse reactions were reported from clinical trials conducted in 558 patients with ovarian cancer (331 received olaparib and 227 received placebo).
SOLO-2
In the SOLO-2 study, the safety of this product was evaluated for maintenance therapy in patients with platinum-sensitive germline breast cancer susceptibility mutations (gBRCAm) ovarian cancer. The study was a placebo-controlled, double-blind study in which 294 patients received either 300 mg of this product (2×150 mg tablets) twice daily (n=195) or placebo tablets twice daily (n=99) until disease progression or intolerable toxicity developed. The median duration of study treatment was 19.4 months for patients receiving this product and 5.6 months for patients receiving placebo. Treatment interruptions due to adverse reactions of any grade occurred in 45% of patients treated with this product compared to 18% in the placebo group, and dose reductions due to adverse reactions occurred in 27% of patients compared to 3% in the placebo group. The most common adverse reactions leading to treatment interruption or dose reduction included anemia (22%), neutropenia (9%), and fatigue/weakness (8%). Treatment with this product resulted in discontinuation in 11% of patients compared to 2% in the placebo group.
Table 1 summarizes the adverse reactions that occurred in at least 20% of patients treated with this product in SOLO-2. Table 2 lists the abnormal laboratory tests that occurred in at least 25% of patients treated with SOLO-2 receiving this product.
Table1 Adverse Reactions in SOLO-2a (≥20%patients treated with olaparib tablets)
Adverse reactions
Olaparib tablets
n=195
Placebo
n=99
1-4Grades
%
Grades 3-4
%
Grades 1-4
%
Grades 3-4
%
Diseases of the blood and lymphatic system
Anemiab
44
20
9
2
Gastrointestinal Disorders
Nausea
76
3
33
0
Vomiting
37
3
19
1
Diarrhea
33
2
22
0
Oral mucositisc
20
1
16
0
Infectious and Infectious Diseases
Nasopharyngitis/upper respiratory tract infection/sinusitis/rhinitis/influenza
36
0
29
0
Systemic disease and various reactions at the site of administration
Fatigue (including weakness)
66
4
39
2
Metabolic and nutritional disorders
Loss of appetite
22
0
11
0
Various musculoskeletal and connective tissue disorders
Arthralgia/Myalgia
30
0
28
0
All types of neurological disorders
Taste Disorders
27
0
7
0
Headache
26
1
14
0
a Graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events (CTCAE) version 4.0
b represents categorical terms that include anemia, decreased erythrocyte pressure volume, decreased hemoglobin, iron deficiency, increased mean cell volume, and decreased red blood cell count
c stands for categorical terms including oral abscess, orofacial ulcer, gingival abscess, gum disease, gum pain, gingivitis, oral ulcer, mucosal infection, mucosal inflammation, oral candidiasis, oral discomfort, oral herpes, oral infection, oral mucosal erythema, oral pain, oropharyngeal discomfort, and oropharyngeal pain
In addition, adverse reactions that occurred in 20% of patients treated with this product in SOLO-2<were neutropenia, rash, cough, dyspepsia, leukopenia, hypomagnesemia, dizziness, thrombocytopenia, elevated serum creatinine, lymphopenia, and edema.
Table2 Abnormal laboratory tests reported in ≥25% of patients with SOLO-2
Laboratory indicatorsa
Olaparib Tablets
nb=195
Placebo
nb=99
1-4Grades
%
Grades 3-4
%
Grades 1-4
%
Grades 3-4
%
Average red blood cell volume increasec
89
–
52
–
Lowered hemoglobin
83
17
69
0
Lowered white blood cell count
69
5
48
1
Decreased lymphocyte count
67
11
37
1
Decreased absolute neutrophil count
51
7
34
3
Elevated serum creatinine
44
0
29
0
Lower platelet count
42
2
22
1
a Patients with laboratory test values of CTCAE grade 1 were allowed to be enrolled in the clinical study.
b Represents the number of people in the safety data set, with data in the table based on each laboratory metric for all evaluable patients.
c Represents the proportion of subjects with a mean erythrocyte volume>upper limit of normal (ULN)
Study19
Among patients treated with olaparib capsules, 35% had treatment interruptions due to adverse effects compared with 10% in the placebo group; 26% required dose reduction compared with 4% in the placebo group; and 6% led to discontinuation compared with 2% in the placebo group.
Table 3 summarizes the adverse reactions that occurred in at least 20% of the patients treated with olaparib in Study 19. Table 4 lists the abnormal laboratory tests that occurred in at least 25% of the patients treated with olaparib in Study 19.
Table3 Adverse Reactions in Study19a< strong>(≥
20% of patients treated with olaparib)
n=195
n=99
%
%
%
%
b represents categorical terms that include anemia, decreased erythrocyte pressure volume, decreased hemoglobin, iron deficiency, increased mean cell volume, and decreased red blood cell count
c stands for categorical terms including oral abscess, orofacial ulcer, gingival abscess, gum disease, gum pain, gingivitis, oral ulcer, mucosal infection, mucosal inflammation, oral candidiasis, oral discomfort, oral herpes, oral infection, oral mucosal erythema, oral pain, oropharyngeal discomfort, and oropharyngeal pain
In addition, adverse reactions that occurred in 20% of patients treated with this product in SOLO-2<were neutropenia, rash, cough, dyspepsia, leukopenia, hypomagnesemia, dizziness, thrombocytopenia, elevated serum creatinine, lymphopenia, and edema.
Table2 Abnormal laboratory tests reported in ≥25% of patients with SOLO-2
nb=195
nb=99
%
%
%
%
b Represents the number of people in the safety data set, with data in the table based on each laboratory metric for all evaluable patients.
c Represents the proportion of subjects with a mean erythrocyte volume>upper limit of normal (ULN)
Study19
Among patients treated with olaparib capsules, 35% had treatment interruptions due to adverse effects compared with 10% in the placebo group; 26% required dose reduction compared with 4% in the placebo group; and 6% led to discontinuation compared with 2% in the placebo group.
Table 3 summarizes the adverse reactions that occurred in at least 20% of the patients treated with olaparib in Study 19. Table 4 lists the abnormal laboratory tests that occurred in at least 25% of the patients treated with olaparib in Study 19.
Table3 Adverse Reactions in Study19a< strong>(≥
| Olaparib capsules n=136 |
Placebo n=128 |
|||
| 1-4Grades % |
Grades 3-4 % |
Grades 1-4 % |
Grades 3-4 % |
|
| Diseases of the blood and lymphatic system | ||||
| Anemiab | 23 | 7 | 7 | 1 |
| Gastrointestinal Disorders | ||||
| Nausea | 71 | 2 | 36 | 0 |
| Vomiting | 35 | 2 | 14 | 1 |
| Diarrhea | 28 | 2 | 25 | 2 |
| Constipation | 22 | 1 | 12 | 0 |
| Systemic disease and various reactions at the site of administration | ||||
| Fatigue (including weakness) | 63 | 9 | 46 | 3 |
| Infectious and Infectious Diseases | ||||
| Respiratory tract infections | 22 | 2 | 11 | 0 |
| Metabolic and nutritional disorders | ||||
| Loss of appetite | 21 | 0 | 13 | 0 |
| All types of neurological disorders | ||||
| Headaches | 21 | 0 | 13 | 1 |
a According to the National Cancer Institute (NCI) CTCAE 4.0 classification
b Categorical terms representing related terms that reflect the medical concept of adverse reactions.
In addition, among patients treated with this product in Study 19,<20% experienced adverse reactions of dyspepsia, oral mucositis, taste disturbance, dizziness, elevated blood creatinine, neutropenia, thrombocytopenia, leukopenia, lymphocytopenia, dyspnea, fever, and edema.
Table4 Laboratories reported by ≥25% of patients in study19 tests were abnormal
| Laboratory indicatorsa | Olaparib capsules nb=136 |
Placebo nb=128 |
||
| 1-4Grades % |
Grades 3-4 % |
Grades 1-4 % |
Grades 3-4 % |
|
| Lowered hemoglobin | 82 | 8 | 58 | 1 |
| Increased mean red blood cell volumec | 82 | – | 51 | – |
| Lowered white blood cell count | 58 | 4 | 37 | 2 |
| Decreased lymphocyte count | 52 | 10 | 32 | 3 |
| Decreased absolute neutrophil count | 47 | 7 | 40 | 2 |
| Elevated serum creatinine | 45 | 0 | 14 | 0 |
| Lower platelet count | 36 | 4 | 18 | 0 |
a Patients with laboratory test values of CTCAE grade 1 were allowed to be enrolled in the clinical study
b Represents the number of people in the safety data set, with data in the table based on each laboratory metric for all evaluable patients.
c Represents the proportion of subjects with a mean erythrocyte volume> upper limit of normal (ULN)
Description of specific adverse reactions
Hematologic Toxicity
Overall anemia and other hematologic toxicities were low grade (CTCAE grade 1 or 2), however, CTCAE grade 3 and higher events were still reported.
The median time to first episode of anemia was approximately 4 weeks (approximately 7 weeks for CTCAE ≥ grade 3 events). Anemia can be controlled by interruption of therapy and dose reduction (see [Dosage]) and, if appropriate, transfusion. In the SOLO2 study, the incidence of anemia was 43.6% (19.5% for CTCAE ≥3), and the incidence of dose interruptions, dose reductions, and discontinuations due to anemia was 16.9%, 8.2%, and 3.1%, respectively; 17.9% of patients treated with olaparib required one or more transfusions. A dose-effect relationship between olaparib and hemoglobin reduction has been demonstrated. The incidence of change (decline) in hemoglobin relative to baseline CTCAE grade ≥ 2 was 20%, absolute neutrophil values were 15%, platelets were 5%, lymphocytes were 30%, and leukocytes were 20% (all percentages are approximate %) in clinical studies with olaparib tablets.
The incidence of an increase in mean red blood cell volume from low or normal values at baseline to above the upper limit of normal was approximately 55%, returning to normal after discontinuation of treatment without any clinically meaningful effects.
Complete blood count testing at baseline and subsequent monthly monitoring is recommended during the first 12 months of treatment, followed by periodic monitoring for clinically meaningful changes in parameters that occur during treatment. Dosing interruptions or reductions and/or further therapy may be required based on these changes (see [Dosage] and [Precautions]).
Other Laboratory Findings
In clinical studies of this product, the incidence of CTCAE ≥ grade 2 elevation in blood creatinine values compared with baseline was approximately 15%. Data from a double-blind placebo-controlled clinical study showed that the median creatinine value was elevated by 23% compared with baseline and remained at that level during treatment, returning to baseline after discontinuation of treatment with no clinically significant sequelae. 90% of patients had a CTCAE grade 0 level at baseline and an additional 10% had a CTCAE grade 1 level at baseline.
Nausea and vomiting
Nausea usually occurs very early in treatment, with the first nausea occurring within the first month of treatment initiation in most patients. Vomiting usually occurs early in treatment, with the first vomiting occurring within two months of treatment initiation in the majority of patients. Nausea and vomiting are reported intermittently in the vast majority of patients and can be controlled with dose interruptions, dose reductions, and/or antiemetic therapy. Prophylactic antiemetics are not required.
[Contraindicated]
Contraindicated in persons with hypersensitivity to the active drug ingredient or any of the excipient ingredients. Discontinue breastfeeding during treatment and for 1 month after the last dose (see [Use in Pregnant and Lactating Women]).
[Precautions]Hematologic Toxicity
Hematologic toxicity, including clinical diagnosis and/or laboratory findings of mild or moderate (CTCAE grade 1 or 2) anemia, neutropenia, thrombocytopenia, and lymphocytopenia, has been reported in patients treated with this product. Patients should not start treatment with this product until hematologic toxicity from prior antineoplastic therapy has recovered (hemoglobin, platelet and neutrophil levels should recover to ≤ CTCAE grade 1). Whole blood cell testing is recommended at baseline for the first 12 months of treatment, followed by monthly monitoring, and thereafter periodic monitoring for clinically significant changes in parameters that occur during treatment (see [Adverse Reactions]).
If a patient develops severe or transfusion-dependent hematologic toxicity, treatment should be interrupted and relevant hematologic testing should be performed. If clinically abnormal blood markers remain after 4 weeks of interruption of this drug administration, bone marrow analysis and/or blood cytogenetic analysis is recommended.
Myelodysplastic Syndrome/Acute Myeloid Leukemia
In clinical studies, including long-term survival follow-up, the incidence of myelodysplastic syndrome/acute myeloid leukemia (MDS/AML) was <1.5% in patients receiving this product as monotherapy. The majority of events had an outcome of death. In patients who developed MDS/AML, the duration of olaparib treatment ranged from <6 months to >2 years, with limited data on longer exposures. All patients had underlying factors contributing to MDS/AML and had received prior platinum-based chemotherapy. Some patients had also been treated with other DNA-damaging drugs as well as radiotherapy. The vast majority of patients were gBRCA 1/2 mutation carriers. Some of these patients have a previous history of tumor or bone marrow dysplasia. If MDS and/or AML is diagnosed during olaparib tablet therapy, it is recommended that olaparib tablet therapy should be discontinued and the patient should be treated appropriately.
Non-infectious Pneumonia
The incidence of noninfectious pneumonia (including events with an outcome of death) in patients receiving monotherapy with this product in clinical studies<1.0%. The reported clinical presentation of noninfectious pneumonia was variable and was influenced by many pathogenic triggers (lung cancer and/or metastatic lung cancer, underlying lung disease, history of smoking, and/or history of prior chemotherapy and radiation therapy). If a patient develops new or worsening respiratory symptoms such as dyspnea, cough and fever, or abnormal chest imaging findings, interrupt treatment temporarily and begin relevant investigations immediately. If a diagnosis of non-infectious pneumonia is confirmed, treatment should be discontinued and the patient treated appropriately.
Embryonic–Fetal Toxicity
Based on the mechanism of action of this product (poly ADP ribose polymerase inhibition) and animal studies, it can cause fetal harm when administered to pregnant females. Preclinical studies in rats have shown adverse effects of olaparib on embryo-fetal survival. At exposures below the recommended human dose of 300 mg twice daily, severe fetal malformations can be induced.
This product should not be taken during pregnancy. If the patient becomes pregnant while taking the drug, the patient should be informed of the potential fetal hazard of this product. It is recommended that women of childbearing age be advised to use effective contraception during treatment and for 6 months after the last dose.
It is recommended that male patients and their female partners of childbearing potential be advised that they must use effective contraception during treatment and for 3 months after the last dose, and that they cannot donate sperm (see [Medications for Pregnant and Lactating Women]).
Interactions with other drugs
Combining this product with a strong or moderately potent CYP3A inhibitor is not recommended (see [Dosage]). If a combination of a potent or intermediate-acting CYP3A inhibitor is necessary, the dose should be reduced (see [DOSAGE AND ADMINISTRATION]).
Combining this product with a potent or intermediate-acting CYP3A inducer is not recommended. The prescribing physician should be aware that the efficacy of this product may be significantly reduced in patients already receiving this product who require treatment with a potent or intermediate-acting CYP3A inducer (see [Drug Interactions]).
Effect on the ability to drive and operate machinery
Studies of the effects of olaparib on the ability to drive and operate machinery have not been performed. However, weakness, fatigue, and dizziness have been reported during treatment with this product, and patients experiencing these symptoms should drive or operate machinery with caution.
Effect onQTInterval
The effect of olaparib on cardiac repolarization was assessed in 119 patients after a single dose of 300 mg and in 109 patients after multiple doses of 300 mg twice daily. No clinically relevant effect of olaparib on the QT interval was found.
[Pregnancy and lactation
[Use in Pregnant and Lactating Women]
Contraception
Women of childbearing potential should not become pregnant at the start of and during treatment with this product, and a pregnancy test is required prior to the start of treatment. Women of childbearing potential must use effective contraception during treatment and for 6 months after the last dose of this product (see [Precautions]). It cannot be excluded that olaparib may reduce exposure to CYP2C9 substrates through enzyme induction and that the efficacy of hormonal contraceptives may be reduced when combined with olaparib. Therefore, other non-hormonal contraceptive measures should be considered during treatment and pregnancy tests should be performed periodically (see [Drug Interactions]).
Based on genotoxicity and animal reproductive toxicity studies of this product, it is recommended that male patients (whose spouses are women of childbearing age or pregnant women) should use effective contraception during treatment and for 3 months after the last dose of olaparib and should not donate sperm (see [Precautions]).
Pregnancy
Animal studies have shown reproductive toxicity, including severe teratogenic effects and effects on embryo-fetal viability in rat tests where maternal systemic exposure was lower than human exposure at therapeutic doses (see [Pharmacologic Toxicology]). There are no data on the use of olaparib in pregnant women, but based on the mechanism of action of olaparib, olaparib tablets should not be used during treatment and for 6 months after the last dose of this product in women of childbearing potential who are pregnant and not using reliable contraception.
Lactation
Animal studies of olaparib secretion into breast milk have not been conducted. It is not known whether olaparib or its metabolites are secreted into human breast milk, and based on the pharmacological profile of this product, it is recommended that breastfeeding be discontinued during treatment with olaparib tablets and for 1 month after the last dose (see [Contraindications]).
Fertility
Clinical data on fertility are not available. Animal studies have shown no effect of study drug on conception, but adverse effects on embryo-fetal survival (see [Pharmacology and Toxicology]).
[Pediatric Dosage]
The safety and efficacy of this product in children and adolescents have not been established; therefore, this product is not indicated for pediatric use.
[Geriatric Use]
In clinical studies enrolling 687 patients with advanced solid tumors receiving olaparib tablets 300 mg twice daily as monotherapy, 146 (21%) patients were ≥65 years of age, including 29 (4%) patients ≥75 years of age, and no patients were ≥85 years of age. There were no significant differences in safety and efficacy data between younger patients and older patients treated with olaparib.
[Drug Interactions]
Pharmacodynamic Interactions
Clinical studies combining this product with other antitumor agents, including those that damage DNA, have shown enhanced and prolonged myelosuppressive toxicity. The recommended monotherapy dose does not apply to its combination with antineoplastic agents with myelosuppressive properties.
No studies have been performed combining olaparib with vaccines or immunosuppressive agents. Therefore, caution should be exercised when the above drugs are co-administered with olaparib tablets and patients should be monitored closely.
Pharmacokinetic Interactions
In vitro studies have confirmed that olaparib is an inhibitor and inducer of CYP3A and an inducer of CYP2B6. Olaparib is predicted to be a weak CYP3A inhibitor in humans. In vitro studies have also shown that olaparib is an inhibitor of uridine diphosphate glucuronyl transferase (UGT) 1A1, breast cancer resistance protein (BCRP), organic anion transport protein (OATP) 1B1, organic cation transport protein (OCT) 1, OCT2, organic anion transport protein (OAT) 3, multidrug and toxic compound efflux transport protein (MATE) 1 and MATE2K inhibitors. The clinical relevance of these findings is unknown. In in vitro studies, olaparib is a substrate for the efflux transporter P-glycoprotein (P-gp) and inhibits P-gp. The potential for olaparib to induce P-gp has not been evaluated.
Drugs that may elevate olaparib plasma concentrations
Olaparib is primarily metabolized by CYP3A. In patients (n=57), combined use of itraconazole, a potent CYP3A inhibitor, increased olaparib AUC by 170%. The medium-acting CYP3A inhibitor fluconazole was predicted to increase the area under the plasma concentration versus time curve (AUC) of olaparib by 121%.
Avoid the combination of potent CYP3A inhibitors such as itraconazole, telithromycin, clarithromycin, ketoconazole, voriconazole, nefazodone, posaconazole, ritonavir, lopinavir/ritonavir, indinavir, saquinavir, nelfinavir, boceprevir, telaprevir, or intermediate-acting CYPA inhibitors such as amprenavir (amprenavir), aripitant, atazanavir, ciprofloxacin, crizotinib, darunavir/ritonavir, diltiazem, erythromycin, fluconazole, fosamprenavir, imatinib, verapamil. Olaparib dose should be reduced if a combination of potent or moderately potent CYP3A inhibitors is necessary.
Avoid grapefruit, grapefruit juice, lime and lime juice during olaparib treatment because these foods contain CYP3A inhibitors.
Drugs That May Reduce Olaparib Plasma Concentrations
In patients (n=22), combined use of rifampin, a potent CYP3A inducer, reduced olaparib AUC by 87%. A predicted reduction in olaparib AUC of approximately 60% with intermediate-acting CYP3A inducers.
Avoid combining strong CYP3A inducers such as phenytoin, rifampin, carbamazepine, and St. John’s wort) or intermediate-acting CYP3A4 inducers such as bosentan, efavirenz, etravirine, modafinil, and nafcillin. Olaparib efficacy may be reduced if mid-acting CYP3A inducers cannot be avoided.
[Overdose]
Clinical signs of overdose with this product have not been identified. A small number of patients taking olaparib tablets at daily doses up to 900 mg for two days had no unintended adverse reactions reported. There is no specific management for overdose of this product. If an overdose occurs, the physician should provide symptomatic and generally supportive treatment.
[Clinical Trials]
Maintenance Therapy for Recurrent Ovarian Cancer
The efficacy of olaparib was evaluated in two randomized, placebo-controlled, double-blind, multicenter studies in patients with recurrent ovarian cancer who achieved remission after platinum-containing therapy.
SOLO2Study (D0816C00002)
The SOLO2 study is a randomized, double-blind, placebo-controlled phase III trial evaluating the safety and efficacy of olaparib as maintenance therapy in patients with platinum-sensitive recurrent (PSR) ovarian, fallopian tube, or primary peritoneal cancer with gBRCA1/2 mutations. Enrolled 295 patients with high-grade plasma or endometrioid PSR ovarian cancer who experienced remission (complete remission [CR] or partial remission [PR]) after completion of platinum-containing chemotherapy were randomized in a 2:1 ratio (196 in the olaparib group and 99 in the placebo group) to receive olaparib (300 mg [2 x 150 mg tablets] twice daily) or placebo until disease progression or development of intolerable toxicity.
Patients who had received two or more platinum-containing regimens were enrolled in the study until disease recurrence > 6 months after completion of the penultimate platinum-based chemotherapy. Patients must not have received prior treatment with olaparib or other PARP inhibitors. Patients are allowed to have received prior treatment with bevacizumab, but the treatment regimen used prior to randomization must not have included bevacizumab.
At baseline, all patients carried deleterious or suspected deleterious gBRCA mutations, detected locally (n=236) or by central Myriad CLIA testing (n=59) and subsequently confirmed by BRACAnalysis® CDx (n=286). Large fragment rearrangements were detected in the BRCA1/2 gene in 4.7% (14/295) of randomized patients.
Demographic and baseline characteristics were essentially similar between the olaparib and placebo groups. The median age in both groups was 56 years. Ovarian cancer in greater than 80% of patients was primary. The most common histologic type was plasmacytic (> 90%), and 6% of patients had endometrioid carcinoma. In the olaparib group, 55% of patients had received only second-line therapy in the past, while 45% received third-line therapy or more. In the placebo group, 61% of patients had received prior second-line therapy only and 39% had received third-line therapy or more. The majority of patients had an ECOG physical status score of 0 (81 %). 60% had a platinum-free interval of > 12 months and 40% had > 6-12 months. 47% of patients responded to platinum-containing chemotherapy in complete remission and 53% in partial remission. Seventeen percent and 20% of patients in the olaparib and placebo groups, respectively, had received prior bevacizumab therapy.
The primary study endpoint was progression-free survival (PFS), as measured by the investigators using the Remission Evaluation Criteria in Solid Tumors (RECIST) 1.1 assessment. Secondary study endpoints included time to second disease progression or death (PFS2); OS (overall survival), time to treatment discontinuation or death (TDT), time to first subsequent anticancer treatment or death (TFST), time to second subsequent anticancer treatment start or death (TSST); and health-related quality of life (HRQoL ).
The study met its primary study endpoint with a statistically significant improvement in investigator-assessed PFS in the olaparib group over the placebo group with a risk ratio (HR) of 0.30 (95% CI 0.22-0.41; p<0.0001; median value in the olaparib group 19.1 months vs. 5.5 months in the placebo group). The investigator-assessed PFS results were supported by blinded independent central imaging assessments (HR= 0.25; 95% CI 0.18-0.35; p<0.0001; median value 30.2 months in the olaparib group vs. 5.5 months in the placebo group). at 2 years, 43% of patients treated with olaparib remained progression-free compared with those treated with placebo on the other hand, only 15%.
See Table 5 and Figure 1 for a summary of the primary study endpoint results for patients with gBRCA1/2m PSR ovarian cancer in SOLO2.
Table5 Primary gBRCA1/2m PSR Ovarian Cancer Patients in SOLO2 Summary of Study Endpoint Results (Investigator Assessment)
Olaparib300 mg tabletsbd
(n=196)
Placebo
(n=99)
PFS(63% maturity)
Number of events: total number of patients (%)
107:196 (55)
80:99 (81)
Median time (months) (95% CI)
19.1 (16.3-25.7)
5.5 (5.2-5.8)
HR (95% CI)a
0.30 (0.22-0.41)
P-value (bilateral)
p<0.0001
a HR=Risk Ratio. Value<1 supports olaparib. Analysis stratified by remission response to prior platinum-based chemotherapy (CR or PR) and time to disease progression in penultimate platinum-containing chemotherapy (>6-12 months and >12 months), using log-rank test, was performed.
bd twice daily; PFS progression-free survival; CI confidence interval.
Figure 1 SOLO2: gBRCA1/2m PSR Kaplan-Meier curve for PFS in ovarian cancer patients (63%maturity – investigator assessment)
Time (months) since randomization
Placebobd
Olaparib300 mg bd
Number of patients at risk
Placebobd
Olaparib300 mg bd
bd twice daily; PFS progression-free survival
The secondary endpoints TFST and PFS2 showed sustained and statistically significant improvement in the olaparib group compared with the placebo group (Table 6).
Table6 SOLO2 ingBRCA1/2m PSR Patients Key Secondary Study Summary of endpoint results
Olaparib300 mg tabletsbd
(n=196)
Placebo
(n=99)
TFST(58% maturity)
Number of events: total number of patients (%)
92:196 (47)
79:99 (80)
Median time (months) (95% CI)
27.9 (22.6-NR)
7.1 (6.3-8.3)
HR (95% CI) a
0.28 (0.21-0.38)
P-value* (bilateral)
p<0.0001
PFS2(40% maturity)
Number of events: total number of patients (%)
70:196 (36)
49:99 (50)
Median time (months) (95% CI)
NR (24.1-NR)
18.4 (15.4-22.8)
HR (95% CI) a
0.50 (0.34-0.72)
P value (bilateral)
p=0.0002
* Uncontrolled multiplicity
a HR=risk ratio. Value <1 supports olaparib. Analyses were performed stratified by remission response to prior platinum-based chemotherapy (CR or PR) and time to disease progression in the penultimate platinum-containing chemotherapy (>6-12 months and >12 months), using log-rank tests.
bd twice daily; NR for attainment; CI confidence interval; PFS2 randomized to time to second disease progression or death; and TFST randomized to time to start of first subsequent treatment or death.
Among patients enrolled in the trial with measurable lesions (target lesions at baseline), the objective remission rate was 41% in the olaparib tablet group compared with 17% in the placebo group. Complete remission occurred in 15.0% of patients treated with olaparib tablets who had a lesion (target lesion or non-target lesion at baseline) at study entry, compared with 9.1% of patients receiving placebo.
Patient self-reported outcome (PRO) data showed no difference between patients in the olaparib group and the placebo group based on the Functional Assessment of Cancer Therapy-Ovarian Cancer (FACT-O) Trial Outcome Index (TOI) relative to baseline change assessment.
SOLO2Study (D0816C00002)< strong>–
China Cohort
Chinese patients were enrolled in the SOLO2 study as a separate cohort, and a total of 32 Chinese patients were randomized to receive either 300 mg (2 x 150 mg tablets) twice daily (n=22) or placebo tablets twice daily (n=10) until disease progression or intolerable toxicity developed. Chinese subjects were tested and confirmed to have a gBRCA mutation by the Shenzhen UW Clinical Laboratory. The median age of patients treated with this product was 49 years (range: 37 to 65 years) and the median age of patients treated with placebo was 46.5 years (range: 33 to 67 years). The ECOG physical status score was 0 in 73% of patients in the treatment group and 50% of patients in the placebo group. 59% of all patients achieved complete remission after the last platinum-containing chemotherapy prior to randomization, and 56% of patients had a time to disease progression of 6-12 months after completion of the penultimate platinum-containing chemotherapy. Approximately 23% of patients in the olaparib group and 20% of patients in the placebo group had received 3 or more prior lines of platinum-containing therapy. In the SOLO2 China cohort, this product significantly improved patient PFS (as assessed by the investigators) compared with placebo, with an HR of 0.44 (95% CI: 0.17-1.19; p = 0.0776; median value in the olaparib group was 13.8 months vs. 5.5 months in the placebo group). This result was consistent with independent imaging assessments. Data on patient survival are not yet mature (only 19% of patients had an event).
Table7 SOLO2Chinese CohortgBRCA1/2m PSR Patients With Ovarian Cancer Summary of results for the primary study endpoint (investigator-assessed)
[Drug Interactions]
Pharmacodynamic Interactions
Clinical studies combining this product with other antitumor agents, including those that damage DNA, have shown enhanced and prolonged myelosuppressive toxicity. The recommended monotherapy dose does not apply to its combination with antineoplastic agents with myelosuppressive properties.
No studies have been performed combining olaparib with vaccines or immunosuppressive agents. Therefore, caution should be exercised when the above drugs are co-administered with olaparib tablets and patients should be monitored closely.
Pharmacokinetic Interactions
In vitro studies have confirmed that olaparib is an inhibitor and inducer of CYP3A and an inducer of CYP2B6. Olaparib is predicted to be a weak CYP3A inhibitor in humans. In vitro studies have also shown that olaparib is an inhibitor of uridine diphosphate glucuronyl transferase (UGT) 1A1, breast cancer resistance protein (BCRP), organic anion transport protein (OATP) 1B1, organic cation transport protein (OCT) 1, OCT2, organic anion transport protein (OAT) 3, multidrug and toxic compound efflux transport protein (MATE) 1 and MATE2K inhibitors. The clinical relevance of these findings is unknown. In in vitro studies, olaparib is a substrate for the efflux transporter P-glycoprotein (P-gp) and inhibits P-gp. The potential for olaparib to induce P-gp has not been evaluated.
Drugs that may elevate olaparib plasma concentrations
Olaparib is primarily metabolized by CYP3A. In patients (n=57), combined use of itraconazole, a potent CYP3A inhibitor, increased olaparib AUC by 170%. The medium-acting CYP3A inhibitor fluconazole was predicted to increase the area under the plasma concentration versus time curve (AUC) of olaparib by 121%.
Avoid the combination of potent CYP3A inhibitors such as itraconazole, telithromycin, clarithromycin, ketoconazole, voriconazole, nefazodone, posaconazole, ritonavir, lopinavir/ritonavir, indinavir, saquinavir, nelfinavir, boceprevir, telaprevir, or intermediate-acting CYPA inhibitors such as amprenavir (amprenavir), aripitant, atazanavir, ciprofloxacin, crizotinib, darunavir/ritonavir, diltiazem, erythromycin, fluconazole, fosamprenavir, imatinib, verapamil. Olaparib dose should be reduced if a combination of potent or moderately potent CYP3A inhibitors is necessary.
Avoid grapefruit, grapefruit juice, lime and lime juice during olaparib treatment because these foods contain CYP3A inhibitors.
Drugs That May Reduce Olaparib Plasma Concentrations
In patients (n=22), combined use of rifampin, a potent CYP3A inducer, reduced olaparib AUC by 87%. A predicted reduction in olaparib AUC of approximately 60% with intermediate-acting CYP3A inducers.
Avoid combining strong CYP3A inducers such as phenytoin, rifampin, carbamazepine, and St. John’s wort) or intermediate-acting CYP3A4 inducers such as bosentan, efavirenz, etravirine, modafinil, and nafcillin. Olaparib efficacy may be reduced if mid-acting CYP3A inducers cannot be avoided.
[Clinical Trials]
The efficacy of olaparib was evaluated in two randomized, placebo-controlled, double-blind, multicenter studies in patients with recurrent ovarian cancer who achieved remission after platinum-containing therapy.
SOLO2Study (D0816C00002)
The SOLO2 study is a randomized, double-blind, placebo-controlled phase III trial evaluating the safety and efficacy of olaparib as maintenance therapy in patients with platinum-sensitive recurrent (PSR) ovarian, fallopian tube, or primary peritoneal cancer with gBRCA1/2 mutations. Enrolled 295 patients with high-grade plasma or endometrioid PSR ovarian cancer who experienced remission (complete remission [CR] or partial remission [PR]) after completion of platinum-containing chemotherapy were randomized in a 2:1 ratio (196 in the olaparib group and 99 in the placebo group) to receive olaparib (300 mg [2 x 150 mg tablets] twice daily) or placebo until disease progression or development of intolerable toxicity.
Patients who had received two or more platinum-containing regimens were enrolled in the study until disease recurrence > 6 months after completion of the penultimate platinum-based chemotherapy. Patients must not have received prior treatment with olaparib or other PARP inhibitors. Patients are allowed to have received prior treatment with bevacizumab, but the treatment regimen used prior to randomization must not have included bevacizumab.
At baseline, all patients carried deleterious or suspected deleterious gBRCA mutations, detected locally (n=236) or by central Myriad CLIA testing (n=59) and subsequently confirmed by BRACAnalysis® CDx (n=286). Large fragment rearrangements were detected in the BRCA1/2 gene in 4.7% (14/295) of randomized patients.
Demographic and baseline characteristics were essentially similar between the olaparib and placebo groups. The median age in both groups was 56 years. Ovarian cancer in greater than 80% of patients was primary. The most common histologic type was plasmacytic (> 90%), and 6% of patients had endometrioid carcinoma. In the olaparib group, 55% of patients had received only second-line therapy in the past, while 45% received third-line therapy or more. In the placebo group, 61% of patients had received prior second-line therapy only and 39% had received third-line therapy or more. The majority of patients had an ECOG physical status score of 0 (81 %). 60% had a platinum-free interval of > 12 months and 40% had > 6-12 months. 47% of patients responded to platinum-containing chemotherapy in complete remission and 53% in partial remission. Seventeen percent and 20% of patients in the olaparib and placebo groups, respectively, had received prior bevacizumab therapy.
The primary study endpoint was progression-free survival (PFS), as measured by the investigators using the Remission Evaluation Criteria in Solid Tumors (RECIST) 1.1 assessment. Secondary study endpoints included time to second disease progression or death (PFS2); OS (overall survival), time to treatment discontinuation or death (TDT), time to first subsequent anticancer treatment or death (TFST), time to second subsequent anticancer treatment start or death (TSST); and health-related quality of life (HRQoL ).
The study met its primary study endpoint with a statistically significant improvement in investigator-assessed PFS in the olaparib group over the placebo group with a risk ratio (HR) of 0.30 (95% CI 0.22-0.41; p<0.0001; median value in the olaparib group 19.1 months vs. 5.5 months in the placebo group). The investigator-assessed PFS results were supported by blinded independent central imaging assessments (HR= 0.25; 95% CI 0.18-0.35; p<0.0001; median value 30.2 months in the olaparib group vs. 5.5 months in the placebo group). at 2 years, 43% of patients treated with olaparib remained progression-free compared with those treated with placebo on the other hand, only 15%.
See Table 5 and Figure 1 for a summary of the primary study endpoint results for patients with gBRCA1/2m PSR ovarian cancer in SOLO2.
Table5 Primary gBRCA1/2m PSR Ovarian Cancer Patients in SOLO2 Summary of Study Endpoint Results (Investigator Assessment)
(n=196)
(n=99)
bd twice daily; PFS progression-free survival; CI confidence interval.
Figure 1 SOLO2: gBRCA1/2m PSR Kaplan-Meier curve for PFS in ovarian cancer patients (63%maturity – investigator assessment)
The secondary endpoints TFST and PFS2 showed sustained and statistically significant improvement in the olaparib group compared with the placebo group (Table 6).
Table6 SOLO2 ingBRCA1/2m PSR Patients Key Secondary Study Summary of endpoint results
(n=196)
(n=99)
a HR=risk ratio. Value <1 supports olaparib. Analyses were performed stratified by remission response to prior platinum-based chemotherapy (CR or PR) and time to disease progression in the penultimate platinum-containing chemotherapy (>6-12 months and >12 months), using log-rank tests.
bd twice daily; NR for attainment; CI confidence interval; PFS2 randomized to time to second disease progression or death; and TFST randomized to time to start of first subsequent treatment or death.
Among patients enrolled in the trial with measurable lesions (target lesions at baseline), the objective remission rate was 41% in the olaparib tablet group compared with 17% in the placebo group. Complete remission occurred in 15.0% of patients treated with olaparib tablets who had a lesion (target lesion or non-target lesion at baseline) at study entry, compared with 9.1% of patients receiving placebo.
Patient self-reported outcome (PRO) data showed no difference between patients in the olaparib group and the placebo group based on the Functional Assessment of Cancer Therapy-Ovarian Cancer (FACT-O) Trial Outcome Index (TOI) relative to baseline change assessment.
SOLO2Study (D0816C00002)< strong>–
| Olaparib Tablets 300 mg Tablets bd (n=22) |
Placebo (n=10) |
|
| PFS(65.6% maturation) | ||
| 14:22 (63.6) | 7:10 (70.0) | |
| Median time (months) (95% CI) | 13.8 (10.1, 16.6) | 5.5 (3.2, 8.4) |
| 0.44 (0.17, 1.19) | ||
| p=0.0776 | ||
a HR=risk ratio, risk ratio vs p-value based on unstratified proportional risk model.
PFS Progression-free survival
Efficacy and safety in Chinese subjects were consistent with those in non-Chinese subjects.
Study19(D0810C00019< em>)
Study 19 is a randomized, double-blind, placebo-controlled phase II trial evaluating the safety and efficacy of olaparib as maintenance therapy in patients with PSR ovarian cancer, including those with tubal or primary peritoneal cancer, after two or more platinum-based regimens. Two hundred and sixty-five patients with PSR-grade plasma ovarian cancer who achieved remission (CR or PR) after completion of platinum-containing chemotherapy were enrolled and randomized 1:1 (136 in the olaparib group and 129 in the placebo group) to receive olaparib capsules 400 mg (8 x 50 mg tablets) twice daily (capsules were not declared available in China) or placebo until disease progression or intolerable toxicity developed. The primary endpoint was PFS as assessed by the investigators using RECIST 1.0 criteria. secondary efficacy endpoints included OS, disease control rate (DCR, i.e., confirmed CR/PR + SD [disease stabilization]), HRQoL, and disease-related symptoms. Exploratory analyses were performed for TFST and TSST.
The study enrolled patients from the completion of penultimate platinum-containing chemotherapy to disease relapse>6 months. Inclusion requirements excluded identified BRCA1/2 mutations (some patients were retrospectively tested for BRCA mutation status). Patients must not have received prior treatment with olaparib or other PARP inhibitors. Patients were allowed to have received prior treatment with bevacizumab, but the treatment regimen used prior to randomization must not have included bevacizumab. No further treatment with olaparib is allowed after disease progression occurs during olaparib treatment.
Patients with BRCA1/2 mutations were identified using local tests or Myriad CLIA comprehensive BRACAnalysis® tests for blood germline testing or tests performed on tumor samples using tests performed by Foundation Medicine. Large fragment rearrangements were detected in the BRCA1/2 gene in 7.4% (10/136) of randomized patients.
Demographic and baseline characteristics were essentially similar between the olaparib and placebo groups. The median age in both groups was 59 years. 86% of patients had primary ovarian cancer. In the olaparib group, 44% of patients had received only second-line therapy in the past, while 56% had received third-line therapy or more. In the placebo group, 49% of patients had received prior second-line therapy only, while 51% had received third-line therapy or more. The majority of patients had an ECOG physical status score of 0 (77%). 60% of patients had a platinum-free interval of > 12 months and 40% had > 6-12 months. 45% of patients had a complete response to platinum-containing chemotherapy and 55% had a partial remission. Six percent and 5% of patients in the olaparib and placebo groups, respectively, had received prior bevacizumab therapy.
The study met the primary study endpoint with a statistically significant improvement in investigator-assessed PFS in the olaparib group compared with the placebo group, with an HR of 0.35 (95% CI 0.25-0.49; p<0.00001; median value of 8.4 months in the olaparib group vs. 4.8 months in the placebo group). At the time of final OS analysis (maturity 79%, data cutoff [DCO] May 09, 2016), the risk ratio for olaparib versus placebo was 0.73 (95% CI 0.55-0.95; p=0.02138 [prespecified significance level not reached< 0.0095]; median value in the olaparib group was 29.8 months and in the placebo group was (27.8 months). In the olaparib-treated group, 23.5% (n=32/136) of patients received treatment for ≥2 years compared with 3.9% (n=5/128) of patients in the placebo group. Despite the limited number of patients, 13.2% (n=18/136) of patients in the olaparib group received treatment for ≥5 years compared with 0.8% (n=1/128) in the placebo group.
A preplanned subgroup analysis found that patients with BRCA1/2 mutations in ovarian cancer (n=136, 51.3%; including 20 patients identified with somatic tumor BRCA1/2 mutations) had the greatest benefit from olaparib monotherapy maintenance treatment. Clinical benefit was also observed in patients with BRCA1/2 wild-type/variants of unknown significance (BRCA1/2 wt/VUS), although to a lesser extent. No multiple testing was available for subgroup analysis.
Please see Table 8 and Figure 2 for a summary of the results of the primary study end points in patients with BRCA1/2 mutations and BRCA1/2 wt/ VUS PSR ovarian cancer in Study 19.
Table8 Study19 inBRCA1/2< strong> mutations andBRCA1/2 wt/VUS PSR Summary of primary study endpoint results in patients with ovarian cancer
| All patientsa | BRCA1/2mutations | BRCA1/2 wt/VUS | ||||
| Olaparib400 mg Capsulebd |
Placebo | Olaparib400 mg Capsulebd |
Placebo | Olaparib400 mg Capsulebd |
Placebo | |
| PFS-DCO 2010Year06Month30< strong>Day | ||||||
| Number of events: total number of patients (%) |
60:136 (44) |
94:129 (73) |
26:74 (35) |
46:62 (74) |
32:57 (56) |
44:61 (72) |
| Median time (months) (95% CI) | 8.4 (7.4-11.5) |
4.8 (4.0-5.5) |
11.2 (8.3-NR) |
4.3 (3.0-5.4) |
7.4 (5.5-10.3) |
5.5 (3.7-5.6) |
| 0.35 (0.25-0.49) | 0.18 (0.10-0.31) | 0.54 (0.34-0.85) | ||||
| p<0.00001 | p<0.00001 | p=0.00745 | ||||
a All patients include the following subgroups: BRCA1/2-mutation, BRCA1/2 wt/VUS and BRCA1/2 status unknown (11 patients with unknown status are not included in the
table not as a separate subgroup).
b HR=risk ratio. The value <1 supports olaparib. Analysis was performed using a Cox proportional risk model for treatment, ethnic origin, platinum sensitivity
and remission after last platinum-based chemotherapy as effectors.
wt (wild type) wild type; VUS (variants of uncertain significance) variants of uncertain significance; bd twice daily; PFS progression-free survival; DCO data cutoff; CI confidence interval; NR not reached.
| Time (months) since randomization |
| Placebobd |
| Olaparib400mg bd |
| Olaparib400mg bd |
| Placebobd |
| Number of patients at risk: |
bd twice daily; DCO data cutoff; FAS full analysis set; PFS progression-free survival
Results of key secondary study endpoints for patients with BRCA1/2 mutations and BRCA1/2 wt/VUS PSR ovarian cancer in Study 19 are summarized in Table 9, and all patients are summarized in Table 9 and Figure 3.
Table9 Study19 inBRCA1/2 mutations andBRCA1/2 wt/VUS PSR Summary of results of key secondary study endpoints in patients with ovarian cancer
| All patientsa | BRCA1/2mutations | BRCA1/2 wt/VUS | ||||
| Olaparib400 mg Capsulebd |
Placebo | Olaparib400 mg Capsulebd |
Placebo | Olaparib400 mg Capsulebd |
Placebo | |
| OS – DCO 2016Year05Month09< strong>Day | ||||||
| Number of events: total number of patients (%) | 98:136 (72) |
112:129 (87) |
49:74 (66) |
50:62 (81)c |
45:57 (79) |
57:61 (93) |
| 29.8 (26.9-35.7) |
27.8 (24.9-33.7) |
34.9 (29.2-54.6) |
30.2 (23.1-40.7) |
24.5 (19.8-35.0) |
26.6 (23.1-32.5) |
|
| 0.73 (0.55-0.95) | 0.62 (0.42-0.93) | 0.84 (0.57-1.25) | ||||
| p=0.02138 | p=0.02140 | p=0.39749 | ||||
| TFST – DCO 2016Year05Month09< strong>day | ||||||
| Number of events: total number of patients (%) | 106:136 (78) |
124:128 (97) |
55:74 (74) |
59:62 (95) |
47:57 (83) |
60:61 (98) |
| Median time (months) (95% CI) |
13.3 (11.3-15.7) |
6.7 (5.7-8.2) |
15.6 (11.9-28.2) |
6.2 (5.3-9.2) |
12.9 (7.8-15.3) |
6.9 (5.7-9.3) |
| 0.39 (0.30-0.52) | 0.33 (0.22-0.49) | 0.45 (0.30-0.66) | ||||
| p<0.00001 | p<0.00001 | p=0.00006 | ||||
* There was no multiple testing strategy for subgroup analysis or for TFST in all patients.
a All patients included the following subgroups: BRCA1/2 mutations, BRCA1/2 wt/VUS, and BRCA1/2 status unknown (11 patients with unknown status in
table not as a separate subgroup).
b HR=risk ratio. The value <1 supports olaparib. Analysis was performed using a Cox proportional risk model for treatment, race, platinum sensitivity, and
remission after last platinum-based chemotherapy as effectors.
In the c BRCA mutation subgroup, approximately one-quarter (14/62; 22.6%) of patients in the placebo-treated group were subsequently treated with PARP inhibitors.
wt (wild type) wild type; VUS (variants of uncertain significance) variants of uncertain significance; bd twice daily; OS overall survival; DCO data cutoff; CI: confidence interval; TFST time from randomization to first subsequent treatment or death
Figure 3 Study 19: Kaplan-Meier curves for OS in FAS (79%Maturity, DCO 2016 05 09)
| Placebo |
| Olaparib400mg bd |
| Olaparib400mg bd |
| Placebo |
| Number of patients at risk: |
| Time since randomization (months) |
bd twice daily; DCO data cutoff; FAS full analysis set; OS overall survival
Patient self-reported outcome (PRO) data showed no difference between patients in the olaparib group and the placebo group, as measured by the rate of improvement and rate of worsening in the Functional Assessment of Cancer Therapy-Ovarian Cancer Total Score (Total FACT-O) Trial Outcome Index (TOI).
[Pharmacology and Toxicology]
Pharmacological effects
Olaparib is an inhibitor of poly ADP ribose polymerase (PARP, including PARP1, PARP2 and PARP3). PARPases are involved in normal cellular functions such as DNA transcription and DNA repair. The results of the trial showed that olaparib inhibited the proliferation of tumor cell lines in vitro and the growth of human tumor-bearing mouse xenografts in vivo, and was effective as monotherapy or after platinum-based chemotherapy. Olaparib administration produced stronger cytotoxic and tumor suppressive effects when cell lines and mouse transplantation tumor models had BRCA-associated defects in DNA damage homologous recombination repair or non-BRCA-associated defects in DNA damage homologous recombination repair associated with platinum-based chemotherapeutic response. In vitro studies suggest that the cytotoxic effects of olaparib may involve inhibition of PARPase activity and increased PARP-DNA complex formation, leading to DNA damage and cancer cell death.
Toxicological studies
Genotoxicity: Olaparib Ames test results were negative; Chinese hamster ovary (CHO) cell chromosome aberration test and rat bone marrow micronucleus test results were positive, and chromosome breakage effects were seen, consistent with the pharmacological effects of olaparib-induced genomic instability, suggesting that olaparib may be genotoxic in humans.
Reproductive toxicity: In a female rat fertility and early embryonic development toxicity assay, oral administration of olaparib at doses up to 15 mg/kg/day (maternal systemic exposure of approximately 7% of human exposure [AUC0-24h] at the clinically recommended dose) from 14 days prior to mating to day 6 of gestation had no effect on No effects on mating or fertility were seen, but increased post-coital abortions were induced.
In a fertility trial in male rats, oral administration of olaparib at doses up to 40 mg/kg/day (systemic exposure of approximately 5% of the clinically recommended human exposure [AUC0-24h]) for at least 70 days had no effect on mating or fertility.
In an embryo-fetal developmental toxicity assay, orally administered olaparib 0.05 and 0.5 mg/kg/day to pregnant rats during the organogenesis phase. Embryo-fetal toxicity was seen at a dose of 0.5 mg/kg/day (maternal systemic exposure of approximately 0.18% of the clinically recommended human exposure [AUC0-24h]) and included increased post-arrival abortion and fusion or absence of the eye (anophthalmia, microphthalmia), vertebrae/ribs (additional ribs or ossification centers, vertebral arches, ribs, and sternum), skull (occipital external fusion) and severe malformations of the diaphragm (hernia). Other abnormalities or variants include incomplete or absent ossification (vertebrae/sternum, ribs, extremities) and abnormalities of vertebrae/sternum, pelvic girdle, lung, thymus, liver, ureter, and umbilical artery. The incidence of ocular, rib, and ureteral abnormalities described above was low when administered at a dose of 0.05 mg/kg/day.
Carcinogenicity: No relevant studies have been performed.
[Pharmacokinetics]
The dosage forms of olaparib include tablets and capsules (capsules have not been declared for marketing in China). The oral bioavailability of the tablet form is higher than that of the capsule form. Population pharmacokinetic analysis has confirmed that steady-state exposure (AUC) was 77% higher after 2 daily doses of 300 mg tablets than after 2 daily doses of 400 mg capsules. geometric means of olaparib AUC and Cmax after a single dose of 300 mg tablets were 42.0 μg*h/mL (n = 204) and 5.8 μg/mL (n = 204), and the geometric mean of steady-state AUC and Cmax were 49.0 μg*h/mL (n = 227) and 7.7 μg/mL (n = 227), respectively, after 2 daily doses of 300 mg tablets. Olaparib PK was time-dependent, with a 15% reduction in steady-state clearance after multiple dosing.
Absorption
Following oral administration of olaparib, absorption was rapid, with median peak plasma concentrations typically reached 1.5 hours after administration. a steady-state AUC mean accumulation ratio of 1.8 was observed after multiple dosing of 300 mg tablets twice daily.
Olaparib systemic exposure (single-dose AUC) increased approximately proportionally with dose when the dose range was 25 mg to 450 mg, with Cmax increasing slightly less than the proportional dose increase in the same dose range.
Co-administration with a high-fat diet resulted in a rate of absorption of olaparib (tmax delayed by 2.5 hours) but did not significantly alter the extent of olaparib absorption (mean AUC increased by approximately 8%).
Distribution
After a single dose of olaparib 300 mg, the mean (± standard deviation) apparent volume of distribution of olaparib was 158 ± 136 L. Olaparib had an in vitro protein binding rate of approximately 82%.
Metabolism
In in vitro studies, CYP3A4/5 has been shown to be the enzyme primarily responsible for olaparib metabolism.
In female patients, 14C-olaparib accounts for the majority (70%) of the radioactivity circulating in plasma after oral administration of prototype olaparib. 14C-olaparib is extensively metabolized, with the prototype drug accounting for 15% and 6% in urine and feces, respectively. Most metabolism is attributable to oxidative reactions with many of the components produced undergoing subsequent glucosinolate or sulfate binding.
Excretion
After a single dose of olaparib 300 mg, the mean plasma terminal half-life (± standard deviation) was 14.9 ± 8.2 hours, and the apparent plasma clearance was 7.4 ± 3.9 L/h.
After 14C-olaparib single administration, 86% of the given radioactivity was recovered within the 7-day collection period, 44% via urine, and 42% via feces. Most material was excreted as metabolites.
Special Populations
Patient age, sex, weight, or race (including Caucasian, Chinese, and Japanese) were not significant covariates in the population pharmacokinetic analysis.
Hepatic impairmentIn a trial of hepatic impairment, when olaparib was given to patients with mild hepatic impairment (Child-Pugh classification A; n=9), there was a 15% increase in mean AUC and a 13% increase in mean Cmax compared with patients with normal hepatic function (n=13). Mild hepatic impairment had no effect on the protein binding of olaparib, so total plasma exposure represents free drug. Data are not available in patients with moderate or severe hepatic impairment.
Renal Impairment
In a trial of renal impairment, compared with patients with normal renal function (CLcr ≥81 mL/min; n=12), patients with mild renal impairment (as defined by the Cockcroft-Gault equation, CLcr = 51-80 mL/min; n=13) taking olaparib had a 24% increase in mean AUC, a mean Cmaxincreased by 15%, and patients with moderate renal impairment (CLcr = 31-50 mL/min; n=13) had a mean AUC and Cmaxincreased by 44% and 26%, respectively, after taking olaparib. There was no evidence of a correlation between the degree of olaparib plasma protein binding and creatinine clearance. No data were available for patients with severe renal impairment or end-stage renal disease (CLcr ≤30 mL/min).
[Storage].
Store below 30°C.
[Packaging]
Aluminum-aluminum blister pack, 56 tablets per box (7 plates)
Aluminum-aluminum blister pack, 112 tablets per box (14 plates)
[Expiration date]
36 months
[Executive Standard]
Imported drug registration standards.
[Approval Number]
Company name: AbbVie Deutschland GmbH & Co. KG
Production Address: Knollstrasse 67061 Ludwigshafen, Germany
Address of China Liaison Office: No. 2 Huangshan Road, Wuxi New District, Jiangsu Province
Postal code: 214028
Quality complaints: 400 828 1755, 800 828 1755
Product information toll-free: 400 820 8116, 800 820 8116
Fax: 021-38723255
Website: www.astrazeneca.com.cn