Approval Date.
Ibovitide for Injection Instructions
Please read the instructions carefully and use under the guidance of your physician
[Drug Name].
Generic Name: Epovetel for Injection
Trade Name: Ekornin
English Name: Albuvirtide for Injection
Hanyu Pinyin: Zhusheyong Aiboweitai
[Ingredients
The main ingredient of this product: Albuvirtide
Chemical name: acetyl-tryptophan-tryptophan-tryptophan-tryptophan-tryptophan-tryptophan-tryptophan-tryptophan-tryptophan-tyroso-(N-{2-[2-(N-(3-cis-butyldiimino-propionyl)amino)ethoxy[ “font-family:Arial”>]Ethoxy}acetyl)lye-leu-isoleu-group-glute-leu-isoleu-glute-glutamyl-silk-glutamyl-tenamyl-glutamyl-glutamyl-glutamyl-lai-tenamyl-glutamyl-glutamyl-glutamyl-leu-amide
Chemical structural formula.
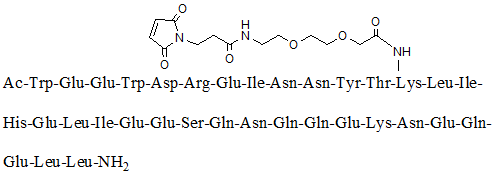
Molecular Formula: C204 H306 N54 O72
Molecular weight: 4666.93
This product has no pharmaceutical excipients.
[Properties
This product is a white or light yellow loose mass or powder.
[Indications
Ebovetide is a human immunodeficiency virus (HIV-1) fusion inhibitor. Only
[Specification
160 mg/vial.
[Dosage].
1. Dosing regimen
Adult and adolescent patients 16 years of age and older: This product is formulated to be administered intravenously at 320 mg/dose once daily on days 1, 2, 3, and 8, and once weekly thereafter.
2. Method of preparation
1) Take 1 vial (bag) of 100 ml of 0.9% sodium chloride injection and discard 12 ml of sodium chloride injection with a disposable syringe and set aside the rest.
2) Take 2 vials of this product and use a 2ml (or 2.5ml) disposable syringe to draw 5% sodium bicarbonate injection into each vial of injectable Epovetel, 1.2ml each, and immediately shake gently until dissolved. The dissolution process takes approximately several minutes. If the phenomenon of solids adhering to the bottle wall occurs during shaking, the bottle needs to be tilted and shaken so that the solution is in full contact with the attached solids. If there are still insoluble particles after 20 minutes, the bottle of drug is discarded and another bottle is taken for preparation.
3) After the drug is completely dissolved, add approximately 6 ml of the alternate 0.9% sodium chloride injection solution to each bottle of injectable Epovetel and shake well. The solution is then withdrawn and added to the spare 0.9% sodium chloride injection vial (bag) and mixed well.
4) The prepared injectable Epovetel solution is to be administered intravenously immediately, not refrigerated or frozen, and should be discarded if not started within 30 minutes after preparation.
Note: The aseptic procedure should be strictly adhered to when dispensing the drug.
3. IV drip dosing rate and precautions
1) Prepare a total of about 90 ml of injectable Epovetel solution and administer it intravenously at a rate of about 2 ml/minute for 45±8 minutes.
2) The formulated solution of Ebovetide for Injection should be colorless or light yellow, clear, transparent, and free of particulate matter. If particulate matter is observed to precipitate prior to or during administration, it should be discarded and not used.
[Adverse Reactions
Ebovitide was used in 170 of the 294 HIV-infected subjects enrolled in 2 phase I, 1 phase II, and 1 phase III clinical trials (interim analysis of results). The primary safety data are based on an ongoing randomized, controlled Phase III clinical trial (TALENT study) with a two-drug combination of epiviride (administered intravenously once weekly) and lopinavir/ritonavir (LPV/r) and a three-drug combination (LPV/r + tenofovir) in the control group. tenofovir (TDF) or zidovudine (AZT) + lamivudine (3TC)) for HIV-1-infected patients already on ART. Interim data analysis summarized safety data for the trial group of 93 cases and the control group of 99 cases treated for 24 to 48 weeks.
Adverse Reactions
Table 1 summarizes the incidence of ≥2% clinical adverse reactions, with diarrhea, headache, dizziness, and rash being common in the trial group.
Table 1 Incidence of adverse reactions with an incidence of ≥2%.
|
|
Trial group (N=93) |
Control group (N=99) |
|||||
|
|
1-2 levels n(%) |
< span style="color:black">3-4 levels n(%) |
< span style="color:black">Total n(%) |
< span style="color:black">1-2 levels n(%) |
< span style="color:black">3-4 levels n(%) |
< span style="color:black">Total n(%) |
|
|
Diarrhea |
7(7.5) |
7(7.5) |
0(0.0) |
7(7.5) |
14(14.1) |
0(0.0) |
14(14.1) |
|
Gastroenteritis |
0(0.0) |
0(0.0) |
0(0.0) |
2(2.0) |
1(1.0) |
3(3.0) |
|
|
Rash |
1(1.1) |
0(0.0) |
1(1.1) |
2(2.0) |
0(0.0) |
2(2.0) |
|
|
Headache |
2(2.2) |
0(0.0) |
2(2.2) |
0(0.0) |
0(0.0) |
0(0.0) |
|
|
Dizziness |
2(2.2) |
0(0.0) |
2(2.2) |
0(0.0) |
0(0.0) |
0(0.0) |
|
|
Hematuria |
0(0.0) |
0(0.0) |
< span style="color:black">0(0.0) |
2(2.0) |
0(0.0) |
2(2.0) |
|
Abnormal results of laboratory tests and ancillary tests
Table 2 summarizes the incidence ≥2% of abnormal laboratory values associated with the study drug. Very common were elevated blood triglycerides and elevated blood cholesterol; common were hyperlipidemia, hypertriglyceridemia, elevated alanine aminotransferase, elevated aspartate aminotransferase, elevated gamma-glutamyl transferase, hyper bilirubinemia, and elevated blood uric acid. The above abnormalities were predominantly mild and moderate elevations (grade 1 to 2), with the incidence of elevated total blood cholesterol higher in the test group than in the control group, and no statistically significant differences between the other abnormal groups.
Table 2 Incidence of laboratory abnormal values with an incidence of ≥2%
|
< span style="color:black">Test group (N=93) |
control group (N=99) |
|||||
|
1-2 levels n(%) |
< span style="color:black">3-4 levels n(%) |
< span style="color:black">Total n(%) |
< span style="color:black">1-2 levels n(%) |
< span style="color:black">3-4 levels n(%) |
< span style="color:black">Total n(%) |
|
|
Elevated blood triglycerides |
22(23.7) |
6(6.5) |
28( 30.1) |
25(25.3) |
4(4.0) |
29(29.3) |
|
Elevated blood cholesterol |
11(11.8) |
1(1.1) |
12(12.9) |
0(0.0) |
2(2.0) |
2(2.0) |
|
Hypertriglyceridemia |
2(2.2) |
0(0.0) |
2(2.2) |
< span style="color:black">0(0.0) |
0(0.0) |
0(0.0) |
|
hyperlipidemia* |
6(6.5) |
1(1.1) |
7(7.5) |
5(5.1) |
0(0.0) |
5(5.1) |
|
Alanine aminotransferase elevation (ALT) |
1(1.1) |
0(0.0) |
< span style="color:black">1(1.1) |
2(2.0) |
0(0.0) |
2( 2.0) |
|
Elevated aspartate aminotransferase (AST) |
1(1.1) |
1(1.1) |
2(2.2) |
0(0.0) |
1(1.0) |
1(1.0) |
|
Elevated gamma-glutamyl transferase (γ -GT) |
0(0.0) |
1(1.1) |
1(1.1) |
1(1.0) |
1(1.0) |
2(2.0) |
|
Abnormal liver function#< /sup> |
3(3.2) |
1(1.1) |
4(4.3) |
2(2.0) |
1(1.0) |
3(3.0) |
|
Elevated blood bilirubin |
2(2.2) |
0(0.0) |
2(2.2) |
1(1.0) |
2(2.0) |
3(3.0) |
|
Elevated blood uric acid |
2(2.2) |
0(0.0) |
2(2.2) |
1(1.0) |
0(0.0) |
1(1.0) |
|
Hepatic steatosis (ultrasound) |
3(3.2) |
0(0.0) |
3(3.2) |
4(4.0) |
0(0.0) |
4(4.0) |
|
Sinus bradycardia (ECG) |
2(2.2) |
0(0.0) |
2(2.2) |
0(0.0) |
0(0.0) |
< span style="color:black">0(0.0) |
*: Elevated blood triglycerides and cholesterol
#: AST, ALT and γ-GT are elevated in 2 or 3 items at the same time
[Contraindicated
It is contraindicated in those who are hypersensitive to this product.
[Precautions
1. This product should be a clear and transparent solution after dissolution.
2. The dissolved solution of this product should be finished in one drip and not used in several times.
[Pregnant and lactating women].
In reproductive toxicity tests in rats and rabbits, intravenous injections of epiviride at 4 and 2 times the adult dose, respectively, showed no toxicity to parental fertility or embryonic development. There are no data from clinical studies on the use of epovetide in pregnant women, and it is not recommended for use in pregnant women.
It is not known whether epiviride is secreted through human breast milk. HIV-infected mothers should not breastfeed their infants to avoid HIV transmission. Breastfeeding women should not breastfeed while on treatment with this product.
[Pediatric Use
The safety and efficacy of this product in pediatric patients are not known.
The safety and efficacy of this product in minors under 16 years of age have not been established.
[Geriatric Use
Clinical studies of this product did not include a sufficient number of subjects aged 65 years and older to confirm whether their effects with this product were different than those of younger subjects.
[Drug Interactions].
In vitro human hepatic microsomal assays showed that epiviride is not a CYP450 enzyme inhibitor and has no significant inhibitory effect on human hepatic microsomal enzyme CYP1A2, 2C8, 2C9, 2C19, 2D6, and 3A4 activities.
In an in vitro combination anti-HIV-1 virus assay, this product was synergistic with zidovudine (AZT) and saquinavir (SQV) and showed additive effects with efavirenz (EFV) and enfuvirtide (T20).
The pharmacokinetic profile of iboviride was not altered by its coadministration with lopinavir/ritonavir (LPV/r), and the in vivo exposure of LPV/r was reduced but no dose adjustment was required.
[Drug overdose].
There is no information on overdose of epojitide in humans. Six HIV-infected patients in clinical trials have received up to a single 640 mg IV dose without drug-related adverse reactions. There is no specific antidote for an epiviride overdose.
[Clinical trials].
Effectiveness of epovetide was evaluated primarily based on a dose-finding study and an ongoing efficacy confirmatory study (TALENT study).
The dose-exploration study was an open, parallel study design evaluating the efficacy and safety of different doses of epovetel in combination with lopinavir/ritonavir (LPV/r) for the treatment of HIV-1-infected patients. Twenty HIV-1-infected patients were enrolled and treated with two doses of epovetel at 160 mg and 320 mg on top of LPV/r, and the effectiveness was evaluated using viral load change as the primary indicator. The results showed that after treatment with epovetel combined with LPV/r, all 20 subjects showed significant decreases in HIV-RNA and varying increases in CD4 levels, and the antiviral effect was significantly better in the 320 mg group than in the 160 mg group.
The confirmatory study (TALENT study) used a multicenter, open, randomized controlled, noninferiority design to evaluate the safety and efficacy of injectable epovalta combined with lopinavir/ritonavir (LPV/r) in HIV-1-infected patients who failed first-line treatment. Subjects were HIV-1-infected and AIDS patients who had already received and failed first-line anti-HIV drug therapy, with 420 planned enrollments and a 48-week treatment cycle during which they received 7 visits. The dosing regimen was: Epovetel injection + LVP/r once daily on days 1, 2, 3, 8 and weekly thereafter for 48 weeks in the trial group; a three-drug combination of lopinavir/ritonavir + tenofovir or zidovudine + lamivudine (LPV/r + TDF or AZT + 3TC) was used in the control group. Primary efficacy indicators: percentage of subjects with HIV-RNA levels <50 copies/ml at week 48 of the study endpoint, evaluated by Snapshot Approach; secondary efficacy indicators: change in log HIV-RNA value after treatment, percentage of HIV-RNA levels <400 copies/mL after treatment, CD4 cell count after treatment change in CD4 cell count after treatment.
The interim data analysis summarizes the effectiveness data from 24 to 48 weeks of treatment in 83 cases in the trial group and 92 cases in the control group.
At 48 weeks of treatment, the FAS set of primary efficacy indicators for the trial and control groups was HIV-RNA <50 copies/ml percent of subjects. =”color:black”>were 80.4% and 66.0%, respectively, with a two-sided 95% CI of -3.0 to 31.9% for the difference between the groups, and the non-inferiority test was passed with a pre-defined non-inferiority threshold of 12%, and the efficacy of the test group was not inferior to that of the control group, as shown in Table 3. the PPS set of test and control groupsHIV-RNA <50 copies/ The percent of subjects was 94.9% and 74.4%, respectively, which was statistically different (P < 0.05), indicating that the efficacy of the test group was better than that of the control group.
Regression analysis (logistic) showed that subject gender, baseline viral load, and CD4 count level had no significant effect on the percentage of HIV-RNA <50 copies/ml at 48 weeks and did not affect treatment efficacy. Ebovetide was effective in controlling viral replication in patients with high viral load infections above 100,000 copies/ml and in those with CD4 cell counts below 100 copies/μl, as shown in Table 3.
Table 3 Results of primary and secondary efficacy indicators (FAS set)
|
Time (week) |
Test group |
control group |
|
|
48 |
80.4% |
66.0% |
|
|
24 |
79.5% |
78.3% |
|
|
<50 copies/ml stratified analysis | |||
|
Baseline HIV-RNA |
|||
|
<100000 copies/ml |
48 |
82.1% |
66.7% |
|
≥100000 copies |
48 |
71.4% |
62.5% |
|
baseline CD4 cell count |
|||
|
<100 units/μl |
48 |
75.0% |
100.0% |
|
≥100/μl |
48 |
81.0% |
|
|
Gender |
|||
|
Male |
48 |
78.8%< /span> |
72.2% |
|
Female |
48 |
84.6% |
50.0% |
|
Subsequent Efficacy metrics |
|||
|
HIV-RNA <400 copies/ml Percentage of subjects (%) |
48 |
84.8% |
74.0% |
|
24 |
89.2% |
82.6% |
|
|
Average change in HIV-RNA from baseline difference (log10 copies/ml) |
48 |
-2.27±0.96 |
-1.77±1.33 |
|
24 |
-2.00±1.01 |
-1.85±1.16 |
|
|
mean change in CD4+ cell count from baseline difference (pcs/μL) |
48 |
159.0±180.3 |
158.7±138.5 |
|
24 |
114.4± 148.4 |
98.7±141.8 |