Approval Date:
Revision Date:
Instructions for Dexmedetomidine Hydrochloride Injection
Please read the instruction manual carefully and use under the guidance of a physician
[Medication Name]
Generic Name: Dexmedetomidine Hydrochloride Injection
English Name:Dexmedetomidine Hydrochloride Injection
Hanyu Pinyin:Yansuan Youmeituomiding Zhusheye
[Composition]
The main ingredient of this product is dexmedetomidine hydrochloride.
Chemical name:(+)-4-(S )-[1-(2,3-dimethylphenyl)ethyl]-1H–imidazole hydrochloride< span style="font-family:equivocal">. < span style="font-family:Times New Roman">
Chemical structure formula:
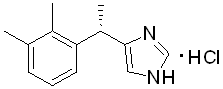
Molecular Formula:C13H16N2-HCl
Molecular weight:236.7
Excipients: sodium chloride, water for injection
[Properties]
This product is a clear, colorless liquid.
[Indications
1. For sedation during tracheal intubation and mechanical ventilation in surgical patients undergoing general anesthesia
2. For sedation of patients starting intubation and ventilated during intensive care treatment, this product should not be infused continuously for more than 24 h.
[Specifications
[Dosage].
Adult dose: 4 μg/ml at 1 μg/kg for 10 min, followed by 0.2 to 0.7 μg/kg/h maintenance infusion. The rate of infusion of the maintenance dose should be adjusted to obtain the desired sedative effect.
Preparation: This product must be diluted to a concentration of 4 μg/ml with 0.9% NaCl solution before administration. 2 ml of this product can be removed and added to 48 ml of 0.9% NaCl injection solution to form a total of 50 ml of solution, shaking gently to mix evenly. Strict aseptic practice must be maintained at all times during the procedure.
The drug should be visually inspected for particulate matter and color change prior to intravenous administration.
Dosage adjustment.
Dose reductions may be required when this product is administered concomitantly with other anesthetics, sedatives, tranquilizers, or opioids due to possible pharmacodynamic interactions (see Drug Interactions).
Patients with hepatic or renal impairment and elderly patients may need to consider reducing the dose administered.
Drug Compatibility.
Because physical compatibility is uncertain, this product should not be given concomitantly with blood or plasma through the same intravenous catheter. It shows incompatibility when given concomitantly with: amphotericin B, diazepam.
Compatibility has been shown when this product is given concomitantly with the following injectable solutions: 0.9% sodium chloride injection, 5% dextrose injection.
Some types of natural rubber have been shown to absorb this product, and synthetic or coated rubber pad delivery devices are recommended.
[Adverse Reactions
Because clinical trials are conducted under many different circumstances, the incidence of adverse reactions observed with one drug in clinical trials cannot be directly compared with another drug and may not reflect the adverse reactions observed in actual clinical use. Foreign studies have reported the use of dexmedetomidine hydrochloride injection to be associated with the following serious adverse reactions.
-
hypotension, bradycardia, and sinus arrest (see Precautions)
-
Transient hypertension (see Precautions)
The most common treatment-related adverse reactions with an incidence greater than 2% have been reported in foreign studies as hypotension, bradycardia, and dry mouth.
The following (including post-marketing conditions) are the incidences of adverse reactions in foreign clinical studies.
Intensive care unit (ICU) patient sedation
Adverse reaction information was obtained from a trial of 1007 patients in the intensive care unit receiving sedation with continuous infusion of dexmedetomidine hydrochloride injection. The mean total dose was 7.4 µg/kg (range: 0.8-84.1), the mean hourly dose was 0.5 µg/kg/hr (range: 0.1-6.0), and the mean infusion time was 15.9 hours (range: 0.2-157.2). The trial population ranged from 17 to 88 years, 43% were ≥65 years old, 77% were male, and 93% were Caucasian. Table 1 shows the incidence >2% of adverse drug reactions. The most common adverse reactions were hypotension, bradycardia, and dry mouth.
Table 1: Adverse reactions with incidence >2% – Subjects sedated in intensive care unit patients Cohort
|
Body systems/adverse reactions |
Dexmedetomidine All treatments N=1007 |
Dexmedetomidine Randomized therapy N=798 |
N=400 |
N=188 |
|
|
n( %) |
n(%) |
n(%) |
n (%) |
||
|
Vascular aspects |
|||||
|
Hypotension |
248 (25%) |
191 (24%) |
48 (12%) |
25 (13%) |
|
|
High blood pressure |
123 (12%) |
101 (13%) |
7 (4%) |
||
|
Gastrointestinal reactions |
|||||
|
Nauseating |
90 (9%) |
73 (9%) |
36 (9%) |
20 (11%) |
|
|
Dry mouth |
35 (4%)< /p> |
22 (3%) |
4 (1%) |
1 (1%) |
|
|
Vomiting |
34 (3%) |
< p>26 (3%) |
21 (5%) |
6 (3%) |
|
|
Heart |
|||||
|
Bradycardia |
52 (5%)< /p> |
36 (5%) |
10 (3%) |
0 |
|
|
Atrial fibrillation |
44 (4%) |
37 (5%) |
13 (3%) |
14 ( 7%) |
|
|
Tachycardia |
20 (2%) |
15 (2%) |
17 (4%) |
2 (1%) |
|
|
Sinus tachycardia > |
6 (1%) |
6 (1%) |
2 (1%) |
4 (2%) |
|
|
Ventricular tachycardia |
4 (0%) |
4 (1%) |
3 (1%) |
9 (5%) |
|
|
Systemic and site of administration symptoms |
|||||
|
Fever |
35 (4%) |
31 (4%) |
15 ( 4%) |
8 (4%) |
|
|
High fever |
19 (2%) |
16 (2%) |
12 (3%) |
0 |
|
|
Chills |
17 (2%) |
14 (2%) |
13 (3%) |
4 (2%) |
|
|
Peripheral edema |
4 (0%) |
2 (0%) |
2 (1%) |
4 (2%) |
|
|
Metabolic and nutritional disorders |
|||||
|
Reduced blood volume |
Reduced blood volume |
31 (3%) |
22 (3%) |
9 (2%) |
9 (5%) |
|
Hyperglycemia |
17 (2%) |
15 (2%) |
7 (2%) |
5 (3%) |
|
|
5 (3%) |
|||||
|
Low blood calcium |
7 ( 1%) |
7 (1%) |
0 |
4 (2%) |
|
|
Acidosis |
6 (1%) |
5 (1%) |
4 (1%) |
4 (2%) |
|
|
Respiratory, thoracic, and mediastinal disorders |
“padding-left: 7px; padding-right: 7px; border-top: none; border-left: none; border-bottom: none; border-right: solid 0.5pt”> | ||||
|
Pulmonary opacity |
29 (3%) |
23 ( 3%) |
13 (3%) |
12 (6%) |
|
|
Pleural leak |
Pleural leak |
23 (2%) |
16 (2%) |
4 (1%) |
12 (6%) |
|
Hypoxia |
16 (2%) |
13 (2%) |
8 (2%) |
5 (3%) |
|
|
5 (3%) |
|||||
|
Pulmonary edema |
9 (1%) |
9 (1%) |
3 (1%) |
5 (3%) |
|
|
Gasping |
4 (0%) |
4 (1%) |
1 (0%) |
4 (2%) |
|
|
Psychiatric symptoms < /td> | |||||
|
Kyoto |
20 (2%) |
16 (2%) |
11 (3%) |
1 (1%) |
|
|
Blood and lymphatic system disorders |
|||||
|
Anemia |
19 (2%) |
18 (2%) |
7 (2%) |
4 (2%) |
|
|
Injuries, poisonings and complications |
|||||
|
Bleeding after drug administration |
15 (2%) |
13 (2%) |
10 (3%) |
7 (4%) |
|
|
Observation |
|||||
|
Decreased urination |
6 (1%) |
6 (1%) |
0 |
4 (2%) |
Procedural sedation.
Adverse reaction information was derived from two trials of procedural sedation in which a total of 318 patients were treated with dexmedetomidine hydrochloride injection. The mean total dose was 1.6 µg/kg (range: 0.5 to 6.7), the mean hourly dose was 1.3 µg/kg/hr (range: 0.3 to 6.1), and the mean infusion time was 1.5 hours (range: 0.1 to 6.2). The trial population ranged from 18 to 93 years of age, 30% of patients were ≥65 years of age, 52% were male, and 61% were Caucasian.
Table 2 shows the incidence >2% of adverse drug reactions. The most common adverse reactions were hypotension, bradycardia, and dry mouth. The footnotes in the table are vital signs indicators reported as predefined criteria for adverse reactions. The incidence of decreased respiratory rate and hypoxia in patients in both trials was similar in both the dexmedetomidine hydrochloride injection administration and control groups.
Table 2: Incidence>2% of adverse reactions – subject population with procedural sedation
|
Body systems/adverse reactions |
Dexmedetomidine Hydrochloride Injection N=318 |
N=113 |
||
|
n( %) |
n(%) |
|||
|
Vascular aspects |
||||
|
Low blood pressure1 |
173 (54%) |
34 (30%) |
||
|
High blood pressure2 |
41 (13%) |
27 (24%) |
||
|
Respiratory, thoracic, and mediastinal disorders > |
||||
|
Respiratory depression5 |
117 (37%) |
36 (32%) |
||
|
Lack of oxygen6 |
7 (2%) |
3 (3%) |
||
|
Slow breathing |
5 (2%) |
5 (4%) |
||
|
< strong>Heart side |
||||
|
Bradycardia3 |
45 (14%) |
4 (4%) |
||
|
Tachycardia4 |
17 (5%) |
19 (17%) |
||
|
Nauseating |
10 (3%) |
2 (2%) |
||
|
Dry mouth |
8 (3%) |
1 (1%) |
- < li>
-
Hypertension is defined in absolute and relative terms as a systolic blood pressure >180 mmHg or more than 30% above the pre-test drug infusion value, or a diastolic blood pressure >100 mmHg.
-
Bradycardia is defined absolutely and relatively as a heart rate <40 beats per minute or less than 30% lower than the pre-infusion value.
-
Tachycardia is defined absolutely and relatively as a number of beats per minute >120 or more than 30% higher than before the infusion value.
-
Absolute and relative definition of respiratory depression as respiratory rate <8 beats per minute or more than 25% lower than baseline.
-
Absence of oxygen was defined absolutely and relatively as SpO2 <90% or a 10% reduction from baseline.
Post-marketing status.
The following adverse reactions were observed after dexmedetomidine hydrochloride injection was approved for marketing. Because these adverse reactions were spontaneously reported from a drug-using population with an as yet unknown sample size, the frequency of occurrence and the determination of causality with the drug cannot be estimated with certainty.
Low blood pressure and bradycardia were the most common adverse reactions after approval of dexmedetomidine hydrochloride injection.
Table 3: Adverse reactions seen after marketing of dexmedetomidine hydrochloride injection
|
Body System |
< strong>Preferences |
|
Full body |
Fever, high fever, decreased blood volume, shallow anesthesia, pain, stiffness |
|
Cardiovascular Systems (Overall) |
Blood pressure fluctuations, heart disease, hypertension, hypotension, myocardial infarction |
|
Central and peripheral nervous system |
Dizziness, headaches, neuralgia, neuritis, speech disorders, tics |
|
Gastrointestinal system |
Abdominal pain, diarrhea, vomiting, nausea |
|
Heart rate and rhythm disorders< |
Irregular heart rate, ventricular arrhythmia, bradycardia, hypoxia, AV block, cardiac arrest, extrasystole, atrial fibrillation, heart block, T-wave inversion, tachycardia, supraventricular tachycardia, ventricular tachycardia |
|
Hepatobiliary system disorders |
Elevated gamma-glutamyl transpeptidase, abnormal liver function, elevated blood bilirubin, elevated alanine aminotransferase, elevated aspartate aminotransferase |
|
Metabolic and nutritional disorders |
Acidosis, respiratory acidosis, hyperkalemia, elevated alkaline phosphatase, thirst, hypoglycemia |
|
Irritation, confusion, delusion, hallucination, fantasy |
|
|
< strong>Red blood cell abnormalities |
Anemia |
|
Kidney disease |
Increased blood urea nitrogen, oliguria |
|
Respiratory system |
Apnea, bronchospasm, dyspnea, hyper carbonic acidemia, hypoventilation, hypoxia, pulmonary congestion |
|
Dermis and appendages |
Increased sweating |
|
Vessels |
Bleeding |
|
Visual impairment |
Flash hallucinations, visual abnormalities |
[Contraindicated
[Precautions
This product should only be used by professionals with medical monitoring equipment. Due to the known pharmacologic effects of this product, patients should be monitored continuously while infusing this product.
Low blood pressure, bradycardia, and sinus arrest
Clinically significant bradycardia and sinus arrest have been reported after administration of this product in healthy young volunteers with high vagal tone or different modes of administration (e.g., rapid intravenous injection or push).
Low blood pressure and bradycardia have been reported in association with this infusion. If treatment is needed, it may include reducing or stopping the infusion of this product, increasing the flow rate of intravenous fluids, elevating the lower extremities, and using drugs that elevate blood pressure. Because this product has the potential to exacerbate vagal stimulation-induced bradycardia, clinicians should be prepared to intervene. Intravenous administration of anticholinergic drugs (eg, glonoprimide, atropine) to reduce vagal tone should be considered. In clinical trials, atropine or glonopin was effective in the treatment of most bradycardic events caused by this product. However, in some patients with significant cardiovascular dysfunction, further resuscitation is required.
Caution should be exercised when giving this product to patients with advanced heart block and/or severe ventricular insufficiency. Because it decreases sympathetic nervous system activity, more hypotension and/or bradycardia may occur in patients with hypovolemia, diabetes mellitus, or chronic hypertension, as well as in elderly patients.
When other vasodilators or negative frequency-acting drugs are given, concomitant administration of this product may have additive pharmacodynamic effects and should be administered with caution.
Transient hypertension
The development of transient hypertension has been observed primarily during loading doses and is associated with the peripheral vasoconstrictive effects of this product. Transient hypertension usually does not require treatment, however a reduced rate of loading infusion may be desirable.
Awakening power
Some patients given this product are observed to be aroused and alert when stimulated. In the absence of other clinical signs and symptoms, this alone should not be considered evidence of ineffectiveness of this product.
Discontinuation Symptoms
Sedation in patients in the intensive care unit: If this product is administered for more than 24 hours and stopped abruptly, it may cause withdrawal symptoms similar to those reported with colistin, another α2 adrenergic agonist. These symptoms include nervousness, agitation, and headache, accompanied or followed by rapid increases in blood pressure and elevated plasma catecholamine concentrations.
Procedural sedation: no withdrawal symptoms after discontinuation of a short-term infusion of this product (<6 hours).
Liver injury
Because the clearance of dexmedetomidine decreases with the severity of hepatic impairment, dose reduction should be considered in patients with hepatic impairment.
Dependence
The potential dependence of dexmedetomidine in humans has not been studied. However, because studies in rodents and primates have demonstrated that dexmedetomidine has similar pharmacological effects to colistin, abrupt discontinuation of this product may produce colistin-like withdrawal symptoms.
[For pregnant and lactating women
Insufficient good clinical studies have been performed in pregnant women. Dexmedetomidine should be used in pregnant women only if the potential benefit outweighs the potential risk to the fetus.
The safety of this product in pregnant women during labor and delivery has not been studied; therefore, it is not recommended during labor and delivery, including cesarean section.
It is not known if this product is secreted into human milk. Radioisotope traced dexmedetomidine is secreted in breast milk after subcutaneous administration to lactating female rats. Because many drugs are secreted in human milk, this product should be used with caution in nursing women.
[Pediatric Use
The safety and efficacy of this product in pediatric patients younger than 18 years of age is unknown. Therefore, it is not recommended for use in these populations.
[Geriatric Use
Dexmedetomidine is known to be excreted primarily through the kidneys, and the drug is at greater risk of adverse reactions in patients with renal impairment. Older patients have reduced renal function, so doses should be chosen carefully in older patients, and monitoring renal function may be useful.
A total of 729 patients ≥65 years of age and 200 ≥75 years of age were included in the sedation trials of intensive care unit patients in foreign clinical studies. The incidence of bradycardia and hypotension was higher in patients over 65 years of age after administration of this product. Therefore, dose reduction should be considered when using this product in patients over 65 years of age. A total of 131 patients ≥65 years of age and 47 ≥75 years of age were enrolled in clinical trials of procedural sedation. The incidence of hypotension was 72% in patients 65 years of age or older, 74% in patients 75 years of age or older, and 47% in patients 65 years of age or older. Therefore the loading dose should be reduced when using this product in patients over 65 years of age, with a recommended infusion of 0.5 µg/kg over 10 minutes.
[Drug Interactions
Anesthetics/sedatives/hypnotics/opioids
Simultaneous administration of this product and anesthetics, sedatives, hypnotics, and opioids may lead to enhancement of their respective pharmacological effects. Foreign studies have reported on the effects of dexmedetomidine hydrochloride with sevoflurane, isoflurane, propofol, alfentanil, and midazolam. There were no pharmacokinetic interactions between dexmedetomidine and sevoflurane, isoflurane, propofol, alfentanil, and midazolam. However, due to possible pharmacodynamic interactions, when given concomitantly, a lower dose of this product or concomitant anesthetics, sedatives, hypnotics, and opioids may be required.
Neuromuscular blockade
In a foreign study of 10 healthy volunteers, 45 minutes of dexmedetomidine hydrochloride administration at a plasma concentration of 1 ng/ml did not result in a significant increase in neuromuscular blockade associated with rocuronium administration.
[Drug overdose
Foreign data show that in a tolerability clinical study, dexmedetomidine hydrochloride was administered to healthy volunteers at or above doses of 0.2 to 0.7 µg/kg/hr, with maximum plasma concentrations approximately 13 times the upper limit of the therapeutic range. The most significant effects observed in the two subjects who reached the highest dose were first-degree AV block and second-degree heart block, followed by spontaneous resolution of AV block and heart block within one minute, with no hemodynamic effects observed.
In a study of patient sedation in the intensive care unit, 5 patients received an overdose of dexmedetomidine hydrochloride. Two of these patients were asymptomatic: one patient received a loading dose of 2 µg/kg for 10 minutes (twice the recommended loading dose) and one patient received a maintenance infusion dose of 0.8 µg/kg/hr. The other two patients who received a loading dose of 2 µg/kg for 10 minutes developed bradycardia and/or hypotension. One patient who received a push dose of undiluted dexmedetomidine hydrochloride loading dose (19.4 µg/kg) experienced cardiac arrest and was successfully treated.
[Pharmacology and Toxicology
Pharmacological effects
Dexmedetomidine is a relatively selective α2-adrenoceptor agonist with sedative effects. Selective effects on α2-adrenoceptors were seen in animals on slow intravenous infusions of dexmedetomidine at 10-300 µg/kg, but at higher doses (≥1000 µg/kg) on both α1– and α2– receptors on slow intravenous infusions or rapid intravenous injections.
Toxicological studies
Genotoxicity
The Ames test for dexmedetomidine and the positive mammalian cell mutation test were negative; the in vitro human lymphocyte chromosome aberration test in rat S9 metabolic activation conditions and the in vivo micronucleus test in NMRI mice were positive, but the in vitro human lymphocyte aberration test in the presence or absence of human S9 metabolic activation conditions was positive. In vitro human lymphocyte chromosome aberration assay in the presence or absence of human S9 metabolic activation, and in vivo micronucleus assay in CD-1 mice were negative.
Reproductive toxicity
Subcutaneous doses of dexmedetomidine up to 54 µg/kg per day (based on µg/m2, which is below the maximum recommended human intravenous dose) were not observed in male and female rats from 10 and 3 weeks prior to mating until mating. fertility effects.
No teratogenic effects were observed with dexmedetomidine at doses up to 200 µg/kg subcutaneously on days 5-16 of gestation in rats and 96 µg/kg subcutaneously on days 6-18 of gestation in rabbits. Based on µg/m2, the rat dose was equivalent to twice the maximum recommended human intravenous dose; based on plasma drug AUC values, the exposure in rabbits was similar to that at the maximum recommended human intravenous dose. Fetal litter toxicity was seen in rats at a dose of 200 µg/kg, as evidenced by increased post-arrival loss and reduced number of surviving live litters. The no-effect dose was 20 µg/kg (based on µg/m2, which is below the maximum recommended human intravenous dose).
Subcutaneous administration of dexmedetomidine to rats from day 16 of gestation to lactation resulted in a reduction in pup weight at doses of 8 and 32 µg/kg (based on µg/m2, below the maximum recommended human intravenous dose); pups in the 32 µg /Embryonic and fetal toxicity was also seen in the F2 generation at 32 µg/kg, but not at 2 µg/kg.
Placental transit was seen in pregnant rats administered subcutaneously with radiolabeled dexmedetomidine.
[Pharmacokinetics
Foreign study data showed that in a study in healthy volunteers (N=10), respiratory rate and oxygen saturation remained within normal limits when the intravenous infusion dose ranged from 0.2 to 0.7 μg/kg/hr, and no respiratory depression was observed.
After intravenous infusion, the pharmacokinetic parameters of dexmedetomidine were as follows: distribution half-life in the rapid distribution phase (t1/2) of approximately 6 min; final un-eliminated half-life (t1/2) of approximately 2 h; steady-state The volume of distribution (Vss) was approximately 118 L. The clearance rate was approximately 39 L/h. The mean body weight used to evaluate clearance was 72 kg.
Intravenous infusions of 0.2 to 0.7 μg/kg/hr until 24 h showed linear pharmacokinetics for dexmedetomidine. Table 4 presents the main pharmacokinetics of dexmedetomidine hydrochloride (after receiving the appropriate loading dose) after 12 and 24 h infusions of 0.17 μg/kg/hr (target concentration of 0.3 ng/ml), 0.33 μg/kg/hr (target concentration of 0.6 ng/ml) and 0.70 μg/kg/hr (target concentration of 1.25 ng/ml), respectively. /ml) following the main pharmacokinetic parameters.
Table 4 Pharmacokinetic parameters (Mean±SD)
|
Parameters |
load infusion time (min)/total infusion time (h) |
||||
|
10 min/ 12h |
10min/24h |
< span style="font-size:12pt">10 min/24h |
35 min/24h |
||
|
Target plasma concentration (ng/ml) and dose (μg/kg/h) of dexmedetomidine |
|||||
|
0.3/0.17 |
0.3/0.17 |
0.6/0.33 > |
1.25/0.70 |
||
|
< span style="font-size:12pt">t*1/2(h) |
1.78 ± 0.30 |
2.22 ± 0.59 |
2.23 ± 0.21 |
2.50 ± 0.61 |
|
|
CL (L/h) |
46.3 ± 8.3 |
43.1 ± 6.5 |
35.3 ± 6.8 < /p> |
36.5 ± 7.5 |
|
|
Vss (L) |
< p>88.7 ± 22.9 |
102.4 ± 20.3 |
93.6 ± 17.0 |
99.6 ± 17.8 |
|
|
Avg Css#(ng/ml) |
0.27 ± 0.05 |
0.27 ± 0.05 |
0.67 ± 0.10 |
1.37 ± 0.20 |
|
*As the reconciled mean and pseudo-fitted standard deviation.
#Avg Css = mean steady-state concentration of dexmedetomidine. (Sampled 2.5 to 9 hours for 12 h infusion and 2.5 to 18 hours for 24 h hour infusion).
Distribution.
The steady-state volume of distribution (VSS) of dexmedetomidine was approximately 118 L. The protein binding of dexmedetomidine was evaluated in the plasma of normal healthy male and female volunteers. Its mean protein binding in different concentration tests was 94%; it was similar in males and females. Dexmedetomidine binding to plasma proteins was significantly reduced in subjects with liver injury compared with healthy subjects.
In vitro studies looked at the potential for replacement of dexmedetomidine binding proteins by fentanyl, ketorolac, theophylline, digoxin, and lidocaine, and no changes in dexmedetomidine plasma protein binding were observed. The possibility that protein binding of phenytoin sodium, warfarin, ibuprofen, ponerol, theophylline, and digoxin was replaced by dexmedetomidine was also investigated in vitro, and the results indicated that no drugs appeared to have protein binding significantly replaced by dexmedetomidine.
Metabolism.
Dexmedetomidine is almost completely biotransformed and rarely excreted in its original form in the urine and feces. Biotransformation includes direct glucosylation and cytochrome P450 enzyme-mediated metabolism. The main metabolic pathways of dexmedetomidine are: direct N-glucosylation to inactive metabolites; lipid hydroxylation (mainly mediated by CYP2A6) to produce 3-hydroxydexmedetomidine, 3-hydroxydexmedetomidine glucosinolate and 3-carboxy-dexmedetomidine; N-methylation of dexmedetomidine to produce 3-hydroxy-N-methyl-dexmedetomidine, 3-carboxy-N-methyl-dexmedetomidine and N-methyl-O-glucuronide dexmedetomidine
Clearance.
The terminal elimination half-life (t1/2) of dexmedetomidine is approximately 2 h, with a clearance rate of approximately 39 L/h. Mass balance studies confirm that an average of 95% of the radioactive material after 9 days of intravenous infusion of radiolabeled dexmedetomidine is recovered from the urine and 4% in the feces. The original form of dexmedetomidine was detectable in urine. Approximately 85% of the radioactive material was excreted in the urine within 24 h after infusion of this product. Segmental separation of the urinary excreted radioactive material was confirmed as N-glucosylated material in 34%. In addition, the fatty hydroxylation products 3-hydroxydexmedetomidine, 3-hydroxydexmedetomidine glucosinolate and 3-carboxylic acid dexmedetomidine accounted for approximately 14%. Dexmedetomidine N-methylation yielded approximately 18% of the 3-hydroxy N-methyl dexmedetomidine, 3-carboxy N-methyl dexmedetomidine, and N-methyl-O-glucosidic acid dexmedetomidine. the N-methyl metabolites themselves were minor circulating components and were not detected in the urine. Approximately 28% of the urinary metabolites were not identified.
Gender.
No gender differences were observed in the pharmacokinetics of dexmedetomidine.
Older patients.
The pharmacokinetic properties of dexmedetomidine do not change with age. There were no differences in the pharmacokinetics of dexmedetomidine in young (18-40 years), middle-aged (41-65 years), and elderly (>65 years) subjects.
Pediatric patients.
The pharmacokinetic properties of dexmedetomidine in pediatric patients have not been studied.
Hepatic impairment.
In subjects with varying degrees of hepatic impairment (Child-Pugh classification A, B, or C), the clearance rate values for dexmedetomidine were lower than in healthy subjects. The mean clearance values in subjects with mild, moderate, and severe hepatic impairment were 74%, 64%, and 53% of those in normal healthy subjects, respectively, and the mean clearance of free drug was 59%, 51%, and 32% of those in normal healthy subjects, respectively.
While administration of this product is required to achieve effectiveness, patients with hepatic impairment may need to consider reducing the dose administered (see Dosage, Precautions).
Renal impairment.
Pharmacokinetics of dexmedetomidine in subjects with severe renal impairment (creatinine clearance: <30 ml/min) (Cmax, Tmax, AUC, t1/2< /sub>, CL, and Vss) were not significantly different compared with healthy subjects. However, the pharmacokinetics of dexmedetomidine metabolites in patients with renal impairment were not evaluated. Because most metabolites are excreted in the urine, long-term infusion in patients with renal impairment is likely to result in metabolite accumulation (see Dosage and Administration).
[Storage]
Store airtight.
[Package]
Neutral borosilicate glass ampoule. 6pcs/ box.
[Expiration date]
24months
[Executive Standard]
[Approval number]
[Manufacturer]
Company Name: Yangtze River Pharmaceutical Group Co.
Manufacturing Address: South Yangzijiang Road, Taizhou City, Jiangsu Province1 family:equals”>No.
Postal Code:225321
Phone Number:400-988-1999
Fax Number: (0523< span style="font-family:equals">)86976161
Net
at:www.yangzijiang.com