Date of approval: 06/05/2015
Revision Date: 23 December 2015
February 09, 2016
February 24, 2017
January 24, 2018
XX/XX/201X
Abiraterone Acetate Tablets Instructions
Please read the instructions carefully and use under the guidance of your physician.
1. [Drug Name]
Generic Name: Abiraterone Acetate Tablets
Trade Name: Zecor® Zytiga®
English Name: Abiraterone Acetate Tablets
Hanyu Pinyin: Cusuan Abitelong Pian
2. [Ingredients]
Main Ingredient: Abiraterone Acetate
Chemical name: 17-(3-pyridyl)-androsta-5,16-diene-3β-acetate
Chemical structure formula: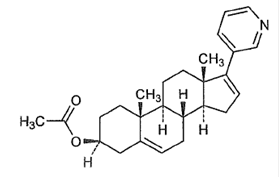
Molecular Formula:C26H33NO2Molecular weight: 391.55
Excipients: lactose monohydrate, sodium cross-linked carboxymethylcellulose, povidone (K29/K32), sodium dodecyl sulfate, microcrystalline cellulose, colloidal silicon dioxide, magnesium stearate
3. [Properties]This product is a white or off-white tablet.
4. [Indications]This product is used in combination with prednisone or prednisolone for the treatment of
Metastatic desmoid-resistant prostate cancer (mCRPC)
Newly diagnosed high-risk metastatic therapy-sensitive prostate cancer (mHSPC), including those that have not received endocrine therapy or have received endocrine therapy for up to 3 months.
5. [Specifications]250mg.
6. [Dosage]
Metastatic desmoid-resistant prostate cancer (mCRPC)
Newly diagnosed high-risk metastatic therapy-sensitive prostate cancer (mHSPC), including those that have not received endocrine therapy or have received endocrine therapy for up to 3 months.
5. [Specifications]250mg.
6. [Dosage]
6.1. Recommended dose
The recommended dose of this product is 1000 mg (4 x 250 mg tablets) taken orally once daily.
This product is used in combination with prednisone or prednisolone 5 mg orally twice daily for the treatment of patients with metastatic debulking resistant prostate cancer (mCRPC).
This product is used in combination with prednisone or prednisolone 5 mg orally once daily for the treatment of newly diagnosed high-risk metastatic treatment-sensitive prostate cancer (mHSPC).
Patients receiving this product should also be receiving concomitant gonadotropin-releasing hormone analogs (GnRHa) or should have undergone bilateral orchiectomy. This product should be taken on an empty stomach at least 1 hour before and 2 hours after a meal (see [Pharmacokinetics]). This product should be swallowed whole with water. Do not break or chew.
Toxicity monitoring during administration
Serum aminotransferases should be tested prior to initiating treatment with this product and every 2 weeks for the first 3 months of treatment and every month thereafter. Blood pressure, serum potassium, and fluid retention should be monitored monthly. However, in patients at significant risk for congestive heart failure, monitoring should be performed every 2 weeks for the first 3 months of treatment and monthly thereafter.
For patients who develop hypokalemia before or during treatment with this product, care should be taken to maintain the patient’s blood potassium level at no less than 4.0 mM.
If patients experience grade 3 and higher toxic events, including hypertension, hypokalemia, edema, or other non-salicorticoid toxic events, treatment should be discontinued and appropriate medical management should be instituted. Treatment with this product should not be restarted until symptoms of toxicity have resolved to Grade 1 or baseline levels.
If a patient experiences a missed dose of this product, prednisone, or prednisolone, treatment should be restarted the following day at the usual dose.
6.2. Principles of dose adjustment in cases of hepatic impairment and hepatotoxicity
Hepatic impairment
No dose adjustment is required for patients with mild hepatic impairment at baseline.
For patients with moderate hepatic impairment at baseline (Child-Pugh class B), the recommended dose of this product should be reduced to 250 mg once daily. A pharmacokinetic study in patients with moderate hepatic impairment at baseline (Child-Pugh class B) showed an approximately 4-fold increase in systemic exposure to abiraterone following a single oral dose of 1000 mg of this product (see [Pharmacokinetics]).
In patients with moderate hepatic impairment, alanine aminotransferase (ALT), aspartate aminotransferase (AST), and bilirubin levels should be monitored prior to initiation of therapy, weekly for the first month, every 2 weeks for the next 2 months, and every month thereafter. If ALT and/or AST are elevated >5 x upper limit of normal (ULN) or total bilirubin is elevated >3 x ULN in patients with moderate hepatic impairment at baseline, discontinue the drug and do not use it again (see [Pharmacokinetics]).
This product should not be used in patients with severe hepatic impairment (Child-Pugh Class C). Another trial analyzed the pharmacokinetics of abiraterone in eight subjects with severe hepatic impairment (Child-Pugh Class C) at baseline and eight healthy control subjects with normal liver function. Subjects with severe hepatic impairment at baseline had a 7-fold increase in systemic exposure (AUC) to abiraterone and a 2-fold increase in exposure to the free drug fraction compared with subjects with normal hepatic function.
6.3. Hepatotoxicity
In patients who experience hepatotoxicity (ALT and/or AST>5×ULN or total bilirubin>3×ULN) during treatment with this product, treatment with this product should be temporarily interrupted and the dose adjusted (see [Precautions]). The dose may be reduced to 750 mg once daily for re-treatment after liver function levels have returned to baseline levels or after AST and ALT ≤ 2.5 x ULN and total bilirubin ≤ 1.5 x ULN. For patients resuming treatment, monitor serum transaminase and bilirubin levels at least every 2 weeks and monthly after 3 months.
If hepatotoxicity occurs again at 750 mg once daily, the dose may be reduced to 500 mg once daily after liver function tests return to baseline or after AST and ALT are ≤2.5 x ULN and total bilirubin is ≤1.5 x ULN.
If hepatotoxicity occurs again at 500 mg once daily, the drug must be discontinued.
In the absence of bile duct obstruction or other causes of concurrent elevation of ALT and total bilirubin, permanent discontinuation is required when a patient develops an ALT > 3 x ULN accompanied by a total bilirubin > 2 x ULN.
6.4. Dose Adjustment in the Setting of Renal Impairment
In patients with renal impairment, no dose adjustment is required (see [Pharmacokinetics]). However, there is no clinical experience in patients with prostate cancer with severe renal impairment, and caution is advised in such patients.
6.5. Dose adjustment in combination with strong CYP3A4 inducers
Avoid combining strong CYP3A4 inducers (e.g., phenytoin sodium, carbamazepine, rifampin, rifabutin, rifapentine, phenobarbital) during treatment with this product. If a combination of strong CYP3A4 inducers is necessary, increase the frequency of dosing to twice daily (e.g., from 1000 mg once daily to 1000 mg twice daily) during the combination period. After discontinuation of the combined strong CYP3A4 inducer, the dose and frequency should be adjusted to the original dose and frequency of administration (see [Drug Interactions]).
Elderly.
See [Geriatric Dosage].
Children or adolescents
The safety and efficacy of this product in children and adolescents have not been established.
7. [Adverse Reactions]
Summary of Safety Profile
This product may cause hypertension, hypokalemia, and fluid retention due to pharmacokinetic effects resulting from the mechanism of action. The most common clinical adverse reactions are peripheral edema, hypokalemia, hypertension, and urinary tract infection. Other important adverse reactions include cardiac disease, hepatotoxicity, fractures, and allergic alveolitis. Usually, adverse reactions to salt corticosteroids can be effectively controlled with management. Combination corticosteroids can reduce the incidence and severity of these adverse drug reactions.
7.1. Clinical Trials
The incidence of adverse reactions observed in clinical trials of different drugs is not directly comparable and does not reflect the incidence of adverse reactions observed in clinical practice because of the varying conditions of clinical trials.
In two randomized, placebo-controlled, multicenter clinical trials (study COU-AA-301 and study COU-AA-302), patients with mCRPC were recruited in which patients in the treatment group received 1000 mg of this drug once daily in combination with prednisone 5 mg twice daily. The control group took placebo in combination with prednisone 5 mg twice daily. A third randomized, placebo-controlled, multicenter clinical trial (study 212082PCR 3011) enrolled patients with high-risk mHSPC. Patients in the treatment group were treated with 1000 mg of this product daily in combination with prednisone (5 mg once daily). Patients in the control group received placebo treatment. In addition, 2 randomized, placebo-controlled trials (Study ABI-PRO-3001 and Study ABI-PRO-3002) were conducted in patients with mCRPC. combined safety data from 2230 patients in 5 randomized controlled trials formed the basis for data in precautions, grade 1 to 4 adverse reactions, and grade 1 to 4 laboratory test abnormalities. In all trials, both groups were required to receive GnRHa or have undergone prior orchiectomy.
In the pooled data, the median duration of treatment was 11 months for abiraterone-treated patients (0.1, 43) and 7.2 months for placebo-treated patients (0.1, 43). The most common (≥10%) adverse drug reactions reported in clinical trials and more common (≥2%) in the abirateron treatment group were fatigue, arthralgia, hypertension, nausea, edema, hypokalemia, hot flashes, diarrhea, vomiting, upper respiratory tract infection, cough, and headache.
The most common (>20%) laboratory test abnormalities reported in clinical trials and more common (≥2%) with abiraterone treatment were anemia, elevated alkaline phosphatase, hypertriglyceridemia, lymphopenia, hypercholesterolemia, hyperglycemia, and hypokalemia.
Grade 3-4 adverse events were reported in 53% of patients in the abiraterone group and in 46% of patients in the placebo group. Discontinuation was reported in 14% of patients in the abiraterone group and 13% of patients in the placebo group. The common adverse events (≥1%) that led to discontinuation of this product and prednisone were hepatotoxicity and cardiac disease.
Deaths related to on-treatment adverse events were reported in 7.5% of patients in the abiraterone group and 6.6% of patients in the placebo group. In the abiraterone group, the most common cause of death was disease progression (3.3%). Other> causes of death reported in 5 patients included pneumonia, cardiac arrest, death (no additional information available), and general physical deterioration.
7.2. Study COU-AA-301: Metastatic desmoid-resistant prostate cancer previously treated with docetaxel chemotherapy
Study COU-AA-301 enrolled 1195 patients with metastatic desmoresistant prostate cancer who had received prior docetaxel chemotherapy. The study specified that a patient without liver metastases could not be enrolled if AST and/or ALT were ≥2.5×ULN. Patients with liver metastases were also ineligible for enrollment if AST and/or ALT>5×ULN. Table 1 shows the incidence of adverse reactions or adverse events of special concern in study COU-AA-301 in which the incidence of adverse reactions increased by ≥2% in the treatment group of this product compared with the placebo group. The median duration of treatment with this product in combination with prednisone was 8 months.
Table 1: Adverse reactions or adverse events requiring special attention that occurred at a ≥2% higher rate in the abiraterone acetate group compared with the placebo group in Study 301
| This product + Prednisone (N = 791) |
Placebo + Prednisone (N = 394) |
|||
| All levels 1 % |
Levels 3-4 % |
All Levels % |
Grade 3-4 % |
|
| 30 26.2 |
4.2 3.0 |
23 23 |
4.1 2.3 |
|
| Systemic disease Edema4 |
27 |
1.9 |
18 |
0.8 |
| Vascular and Lymphatic Vessel Disease Hot flashes Hypertension |
19 8.5 |
0.3 1.3 |
17 6.9 |
0.3 0.3 |
| Gastrointestinal Disorders Diarrhea Indigestion |
18 6.1 |
0.6 0 |
14 3.3 |
1.3 0 |
| Infections and Infectious Diseases Urinary tract infections Upper respiratory tract infections |
12 5.4 |
2.1 0 |
7.1 2.5 |
0.5 0 |
| Respiratory, thoracic and mediastinal disease Cough |
11 |
0 |
7.6 |
0 |
| Kidney and urinary tract disorders Urinary frequency Nocturia |
7.2 6.2 |
0.3 0 |
5.1 4.1 |
0.3 0 |
| All types of injuries, poisoning and surgical complications Fractures5 |
5.9 |
1.4 |
2.3 |
0 |
| Heart organ disease Cardiac arrhythmia6 Chest pain or chest discomfort7 Heart failure8 |
7.2 3.8 2.3 |
1.1 0.5 1.9 |
4.6 2.8 1.0 |
1.0 0 0.3 |
| 1 Adverse events are graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE) version 3.0. 2 Includes the terms: arthritis, arthralgia, joint swelling, and joint stiffness. 3 Includes the terms: muscle spasm, skeletal muscle pain, myalgia, musculoskeletal discomfort, and skeletal muscle tonus. 4 Includes the terms: edema, peripheral edema, concussive edema, and generalized edema. 5 Includes all fractures except pathologic fractures. 6 Includes the terms: arrhythmia, tachycardia, atrial fibrillation, supraventricular tachycardia, atrial tachycardia, ventricular tachycardia, atrial flutter, bradycardia, complete atrioventricular block, conduction disturbance, and bradyarrhythmia. 7 Includes the terms: angina pectoris, chest pain, and unstable angina pectoris. Myocardial infarction or ischemia was reported more frequently in the placebo group than in the drug-treated group (1.3% and 1.1%, respectively). 8 Includes the terms: heart failure, congestive heart failure, left ventricular dysfunction, cardiogenic shock, cardiomegaly, cardiomyopathy, and decreased ejection fraction. |
||||
Table 2 shows the laboratory test abnormalities of interest in study COU-AA-301.
Table 2: Laboratory test abnormalities of interest in study COU-AA-301
| This product + Prednisone (N = 791) | Placebo + Prednisone (N = 394) | |||
| All levels% | Grade 3-4% | All Levels% | Grade 3-4% | |
| 63 | 0.4 | 53 | 0 | |
| AST elevation | 31 | 2.1 | 36 | 1.5 |
| hypokalemia | 28 | 5.3 | 20 | 1.0 |
| hypophosphatemia | 24 | 7.2 | 16 | 5.8 |
| ALT elevation | 11 | 1.4 | 10 | 0.8 |
| Elevated total bilirubin | 6.6 | 0.1 | 4.6 | 0 |
.
7.3. Study COU-AA-302: Chemotherapy-naïve metastatic debulking-resistant prostate cancer
Study COU-AA-302 enrolled 1088 patients with previously untreated metastatic desmoresistant prostate cancer without chemotherapy. Patients with concomitant liver metastases were excluded from the study, and patients with AST and/or ALT ≥2.5×ULN were also ineligible for enrollment. Table 3 shows the adverse reactions that occurred in ≥5% of patients in the treatment arm of study COU-AA-302 and had an increased incidence of ≥2% compared to the placebo arm. The median duration of treatment with this product in combination with prednisone was 13.8 months.
| This product + Prednisone (N = 542) |
Placebo + Prednisone (N = 540) |
|||
| All levels 1 % |
Levels 3-4 % |
All Levels % |
Grade 3-4 % |
|
| 39 25 8.7 |
2.2 0.4 0.6 |
34 21 5.9 |
1.7 1.1 0.2 |
|
| Musculoskeletal and connective tissue disorders Joint swelling/discomfort3 Groin pain |
30 6.6 |
2.0 0.4 |
25 4.1 |
2.0 0.7 |
| Gastrointestinal Disorders Constipation Diarrhea Indigestion |
23 22 11 |
0.4 0.9 0.0 |
19 18 5.0 |
0.6 0.9 0.2 |
| Vascular and lymphatic vascular disease Hot flashes Hypertension |
22 22 |
0.2 3.9 |
18 13 |
0.0 3.0 |
| Respiratory, thoracic and mediastinal disease Cough Dyspnea |
17 12 |
0.0 2.4 |
14 9.6 |
0.2 0.9 |
| Psychiatric disorders Insomnia |
14 |
0.2 |
11 |
0.0 |
| All types of injuries, poisonings and surgical complications Contusions Falls |
13 5.9 |
0.0 0.0 |
9.1 3.3 |
0.0 0.0 |
| Infections and Infectious Diseases Upper respiratory tract infections Nasopharyngitis |
13 11 |
0.0 0.0 |
8.0 8.1 |
0.0 0.0 |
| Kidney and urinary tract disorders Hematuria |
10.3 |
1.3 |
5.6 |
0.6 |
| Dermatologic and subcutaneous tissue disorders Rashes |
8.1 |
0.0 |
3.7 |
0.0 |
| 1 Adverse events are graded according to NCI CTCAE version 3.0. 2 Includes the terms: peripheral edema, concussive edema, and systemic edema. 3 Includes the terms: arthritis, arthralgia, joint swelling, and joint stiffness. |
||||
Table 4 shows laboratory test abnormalities that occurred at a rate of 15% or more in study COU-AA-302 and at a higher rate (>5%) in the Benadryl-treated group than in the placebo group.
Table 4.
Table 4: Abnormal laboratory tests that occurred at an incidence of >15% in the abiraterone acetate group and at a higher rate of >5% than the placebo group in study COU-AA-302
| This product + prednisone (N = 542) | Placebo + prednisone (N = 540) | |||
| Grade 3-4 % |
All levels % |
Grade 3-4 % |
||
| 38 |
8.7 |
32 |
7.4 |
|
| Blood biochemistry Hyperglycemia1 Elevated ALT Elevated AST Hypernatremia Hypokalemia |
57 42 37 33 17 |
6.5 6.1 3.1 0.4 2.8 |
51 29 29 25 10 |
5.2 0.7 1.1 0.2 1.7 |
| 1 based on non-fasting blood test | ||||
7.4. Study 212082PCR 3011: Treatment of patients with high-risk mHSPC
Study 212082PCR 3011 enrolled 1199 patients with newly diagnosed high-risk mHSPC who had not previously received cytotoxic chemotherapy. Patients with concomitant liver metastases were excluded, and patients with AST and/or ALT ≥2.5 times ULN were also ineligible for enrollment. All patients received GnRHa during the trial or had previously undergone bilateral orchiectomy. The median duration of treatment with this product in combination with prednisone was 24 months.
Table 5 shows the adverse reactions that occurred in ≥5% of patients in the Benadryl combined with prednisone group and whose incidence increased by ≥2% compared with that in the placebo combined with prednisone group.
| Table5: Study 212082PCR 3011 Adverse reactions in the abiraterone acetate group that occurred at ≥5% and were ≥2% higher than the placebo group1 | ||||||||
| This product in combination with prednisone (N=597) | Placebo (N=602) |
|||||||
| System/Organ Classification | All levels2 | Levels 3-4 | All Levels | Level 3-4 | ||||
| Adverse effects | % | % | % | % | ||||
| Vascular and Lymphatic Vessel Disease | ||||||||
| High blood pressure | 37 | 20 | 13 | 10 | ||||
| Hot flashes | 15 | 0.0 | 13 | 0.2 | ||||
| Metabolic and nutritional disorders | ||||||||
| Hypokalemia | 20 | 10 | 3.7 | 1.3 | ||||
| All types of inspections | ||||||||
| ALT elevation3 | 16 | 5.5 | 13 | 1.3 | ||||
| AST elevation3 | 15 | 4.4 | 11 | 1.5 | ||||
| Infectious and Infectious Diseases | ||||||||
| Urinary tract infections | 7.0 | 1.0 | 3.7 | 0.8 | ||||
| Upper respiratory tract infection | 6.7 | 0.2 | 4.7 | 0.2 | ||||
| All types of neurological disorders | ||||||||
| Headaches | 7.5 | 0.3 | 5.0 | 0.2 | ||||
| Respiratory, thoracic and mediastinal disease | ||||||||
| cough4 | 6.5 | 0.0 | 3.2 | 0 | ||||
| 1 All patients received GnRHa or underwent orchiectomy. 2 Adverse events were graded according to CTCAE version 4.0. 3 Reported as an adverse event or adverse reaction. 4 Including cough, cough sputum, upper respiratory cough syndrome Note: The abnormal laboratory tests listed in Table 6 are defined according to the values reported on the test; an abnormal laboratory test will be reported as an adverse event when, in the opinion of the investigator, it is clinically significant and requires administration of concomitant medication or adjustment of study medication, as listed in Table 5. |
||||||||
Table 6 shows the abnormal laboratory test events that occurred at a rate of 15% or more in study 212082PCR 3011 and at a higher rate (>5%) in the Benzedrine combined with prednisone treatment group than in the placebo group.
7.5. Table 6: Abnormal laboratory tests in the report form for the treatment group of this product in study 212082PCR 3011>15% of patients |
||||
| This product combined with prednisone (N=597) |
Placebo (N=602) |
|||
| Abnormal laboratory tests | Grade 1-4 % |
Grade 3-4 % |
Grade 1-4 % |
Grade 3-4 % |
| 20 | 4 | 14 | 1.8 | |
| Clinical Biochemistry | ||||
| 30 | 9.6 | 6.7 | 1.3 | |
| ALT elevation | 46 | 6.4 | 45 | 1.3 |
| Elevated total bilirubin | 16 | 0.2 | 6.2 | 0.2 |
7.6. Note: Abnormal laboratory tests, as listed in Table 6, are defined based on the value of the test report; an adverse event will be reported when the investigator believes the laboratory test abnormality is clinically significant and requires administration of concomitant medication or adjustment of study medication, as listed in Table 5.
.
7.7. Description of Significant Adverse Reactions:
Cardiovascular Adverse Reactions.
Phase 3 studies (studies COU-AA-301 and ABI-PRO-3001, studies COU-AA-302 and ABI-PRO-3002, and study 212082PCR 3011) excluded patients with uncontrolled hypertension and clinically significant cardiac disease, the latter including myocardial infarction or arterial thrombosis within the previous 6 months, severe or unstable angina, NYHA-defined class III or IV heart failure (study COU-AA-301 and ABI-PRO-3001) or class II-IV heart failure (study 212082PCR 3011, study COU-AA-302 and ABI-PRO-3002) or cardiac ejection fraction<50%. All enrolled patients (including active drug-treated and placebo-treated patients) received concomitant ADT with primary application of GnRHa, which is associated with diabetes, myocardial infarction, cerebrovascular accident, and sudden cardiac death.
In pooled data from five randomized, placebo-controlled clinical studies, the incidence of heart failure was higher in the Benadryl-treated group than in the placebo group (2.6% vs. 0.9%). Grade 3-4 heart failure occurred in 1.3% of patients in the placebo group, resulting in discontinuation of therapy in 5 patients and death in 4 patients. Grade 3-4 heart failure occurred in 0.2% of patients in the placebo group. Two deaths due to heart failure occurred in the placebo group, and there were no treatment discontinuation events.
In the above pooled data, the vast majority of reported arrhythmias were of grade 1-2. There was one arrhythmia-related death and three sudden deaths in the placebo group and five related deaths in the placebo group. There were 7 deaths due to cardiac arrest in the treatment group (0.3%) and 2 in the placebo group (0.1%). There were 3 deaths due to myocardial ischemia or myocardial infarction in the placebo group and 3 in the placebo-treated group.
The following are discussed in detail in [Precautions] of the instruction manual.
- Hypertension, hypokalemia, and fluid retention due to salt corticosteroid overdose
- Adrenocortical insufficiency
- Hepatotoxicity
- Food can increase exposure to this product
Postmarketing Experience
The following additional adverse reactions have been identified during the post-marketing approval use of this product based on spontaneous reports. The frequencies are as follows: uncommon ≥1/1000 and < 1/100, rare ≥1/10,000 and < 1/1000.
Systemic organ classification: Respiratory, thoracic and mediastinal disease
Rare: allergic alveolitis
Systemic organ classification: various musculoskeletal and connective tissue disorders
Unusual: rhabdomyolysis, myopathy
Systemic organ classification: diseases of the hepatobiliary system
Rare: fulminant hepatitis, acute liver failure
Reporting of suspected adverse reactions
It is important to report suspected adverse reactions after a drug has received marketing authorization. This allows for ongoing monitoring of the benefit/risk balance of the drug. Healthcare professionals are required to report any suspected adverse reactions through the National Adverse Reaction Reporting System.
8. [Contraindicated]
- Contraindicated in persons with hypersensitivity reactions to the active ingredient or excipients of this product.
- Contraindicated in women who are pregnant or at risk of pregnancy.
- Contraindicated in patients with severe hepatic impairment (Child-Pugh Class C).
9. [Caution]
9.1. Hypertension, hypokalemia, and fluid retention due to salt corticosteroid overdose
Hypertension, hypokalemia, and fluid retention may be caused by elevated levels of salicorticoids due to the inhibitory effect of this product on CYP17. Monitor patients for hypertension, hypokalemia, and fluid retention at least monthly. Hypertension should be controlled and hypokalemia corrected prior to and during treatment with this product.
Based on pooled data from four placebo-controlled trials using prednisone 5 mg twice daily in combination with abiraterone acetate 1000 mg once daily, grade 3-4 hypokalemia was found in 4% of patients in the treatment group and 2% of patients in the placebo group. Grade 3-4 hypertension was found to occur in 2% of patients in each treatment group and grade 3-4 fluid retention was found to occur in 1% of patients in each treatment group.
In the 212082PCR 3011 study, using prednisone 5 mg once daily in combination with abiraterone acetate 1000 mg once daily, grade 3-4 hyperkalemia was found in 10% of patients in the treatment group and 1% in the placebo group, and grade 3-4 hypertension was observed in 20% of patients and 10% of patients in the placebo group. Grade 3-4 fluid retention occurred in 1% of patients in all treatment groups.
The use of this product should be closely monitored in patients with elevated blood pressure, low potassium and fluid retention that may lead to exacerbation of the underlying condition, such as those with heart failure, recent myocardial infarction, cardiovascular disease or ventricular arrhythmias. Patients with left ventricular ejection fraction (LVEF) <50% or NYHA class III or IV heart failure (study COU-AA-301) or NYHA class II to IV heart failure (study COU-AA-302 and study 212082PCR3011) were excluded from clinical studies and the safety of this product for use in these patients is uncertain ( see [Clinical Trials]).
9.2. Adrenocortical insufficiency
Pooled data from five randomized, placebo-controlled clinical trials showed that the incidence of adrenocortical insufficiency was 0.3% and 0.1% in the 2230 patients in the Benadryl-treated group and 1763 patients in the placebo group, respectively. Adrenocortical insufficiency has been reported in patients treated with this product in combination with prednisone upon discontinuation of daily steroids and/or concomitant infections or stressful conditions. Monitor for signs and symptoms of adrenocortical insufficiency, particularly in patients who discontinue prednisone, lower the prednisone dose, or develop an abnormal stress state. Adverse reactions associated with salt corticosteroid overdose resulting from this treatment may mask signs and symptoms of adrenocortical insufficiency. Perform appropriate tests as clinically indicated to confirm the diagnosis of adrenocortical insufficiency. Corticosteroid doses may have to be increased before, during, and after the onset of a stressful situation.
9.3. Hepatotoxicity
Pooled data from five randomized clinical trials showed that grade 3/4 ALT or AST elevations (at least 5 x ULN) occurred in 6% of the 2230 patients treated with this product, usually in the first 3 months after treatment initiation. Patients with elevated baseline ALT or AST were more likely to have elevated liver function parameters than patients with normal baseline liver function. Approximately 1.1% of the 2230 patients in the treatment group discontinued treatment due to elevated ALT and AST or abnormal liver function. No deaths due to hepatotoxicity clearly associated with this product were reported in these clinical trials. This product must be taken on an empty stomach. Fast for at least 2 hours before and 1 hour after dosing. The Cmax and AUC0-∞ (exposure) of abiraterone increased to 17-fold and 10-fold, respectively, with a single dose of this product taken with a meal compared with a single dose taken on an empty stomach. The safety of the increased exposure resulting from multiple concomitant doses of this product with food has not been evaluated (see [Adverse Reactions] and [Pharmacokinetics]). Patients with advanced metastatic prostate cancer (desmoresistant prostate cancer) may experience decreased bone mineral density. This effect is enhanced by the combination of this product with glucocorticoids. Patients with prostate cancer who have been previously treated with ketoconazole may have a lower remission rate. The use of glucocorticoids increases the risk of hyperglycemia, so blood glucose should be measured frequently in patients with diabetes. Several myopathic events have been reported in patients treated with this product. Some patients developed rhabdomyolysis accompanied by renal failure. Most cases occurred within the first month of the treatment period and recovered after discontinuation of the drug. This product should be used with caution in patients treated with a combination of drugs known to be associated with myopathy/rhabdomyolysis. The safety and efficacy of this product in combination with cytotoxic chemotherapy has not been established. This product contains lactose. Patients with rare genetic problems such as galactose intolerance, Lapp (lapp) lactase deficiency, or glucose-galactose absorption disorder should not take this product. This product also contains sodium in excess of 1.18 mmol (or 27 mg) per 4-tablet dose. Patients with restricted sodium intake should be considered. Men with metastatic desmoid-resistant prostate cancer (including those being treated with this product) may be at risk for anemia and sexual dysfunction. In a multicenter open single-arm clinical trial, 33 patients with mCRPC received 1000 mg of this product once daily 1 hour before or 2 hours after a meal, in combination with prednisone 5 mg twice daily. There was no significant change in QTc interval from baseline until day 2 of cycle 2 (e.g. > 20ms). However, due to limitations of the clinical trial design, it cannot be completely excluded that this product may slightly prolong the QTc interval (e.g. <10ms). This product is not indicated for use in women. Based on its mechanism of action and the results of animal studies, this product is contraindicated in women who are pregnant or at risk of pregnancy because it may cause fetal damage and may cause pregnancy termination. This product is not indicated for use in women. It is uncertain whether this product is secreted into breast milk and the effect of this product on lactation and on breastfed infants. It is not known if abiraterone or its metabolites are present in semen. If the patient has sex with a pregnant woman, condoms are required. If the patient has sex with a woman of childbearing age, condom use is required along with another effective form of contraception. Based on animal studies, this product may impair fertility in men of reproductive age.
Serum transaminases (ALT and AST) and bilirubin levels were monitored before initiation of treatment, every 2 weeks for the first 3 months after initiation of treatment, and monthly thereafter. For patients receiving a low dose of 250 mg for moderate hepatic impairment at baseline, monitor ALT, AST and bilirubin levels prior to initiation of therapy, once weekly for the first month of therapy, once every 2 weeks for the next 2 months, and once monthly thereafter. Serum total bilirubin, AST and ALT levels should be monitored promptly if clinical signs or symptoms suggestive of hepatotoxicity are present. If AST, ALT or bilirubin are elevated from baseline values, the frequency of monitoring should be increased. Once AST or ALT rises above 5×ULN or bilirubin rises above 3×ULN, the product should be temporarily discontinued and liver function should be closely monitored.
Treatment with this product at low dose levels should be resumed only after liver function tests have returned to the patient’s baseline level or after AST and ALT are ≤ 2.5 x ULN and total bilirubin is ≤ 1.5 x ULN (see [DOSAGE]). If a patient develops severe hepatotoxicity (AST or ALT ≥ 20 x ULN) at any time during treatment, treatment with this product should be discontinued and should not be reintroduced. Post-marketing reports of acute liver failure, fulminant hepatitis, some of which were fatal, have been rare (see [Adverse Reactions]).
9.4. Food may increase exposure to this product
9.5. Bone mineral density
9.6. Prior ketoconazole use
9.7. Hyperglycemia
9.8. Skeletal muscle reactions
9.9. Combination chemotherapy treatment
9.10. Excipient Intolerance
9.11. Other Potential Risks
Keep out of the reach of children.9.12. QT interval
Effects on driving and ability to operate machines
This product has no or negligible effect on the ability to drive and operate machinery.10. [Use in pregnant and lactating women]
10.1. Pregnancy
No human data are available on the use of this product in pregnant women. In animal reproduction studies, oral administration of abiraterone acetate to pregnant rats during the organogenesis phase was associated with effects on fetal litter development when maternal exposure was approximately ≥0.03 times the exposure (AUC) at the recommended human dose.10.2. Lactation
10.3. Contraception
Based on the results of animal reproduction testing and their mechanism of action, men are advised to use effective contraception during treatment with this product and for 3 weeks after the last dose if their partner is a woman of childbearing age.10.3.1. Fertility
11. [Pediatric Use]The effectiveness and safety of this product for use in children has not been established.
12. [Geriatric Use]
Of the patients treated with this product in clinical trials, 70% were 65 years of age or older, while 27% were 75 years of age or older. No overall differences in safety and efficacy were observed between older and younger patients. No other clinical reports have confirmed a difference in response to this product between older and younger patients, but a higher sensitivity in older patients cannot be excluded.
13. [Drug Interactions]
Interactions with other drugsPotential effects of other drugs on abiraterone exposureBased on in vitro data, this product is a substrate for CYP3A4. In a clinical study of pharmacokinetic interactions in healthy subjects who first received the potent CYP3A4 inducer rifampicin administered at 600 mg daily for 6 days, followed by a single dose of 1,000 mg of this product, the mean abiraterone plasma AUC∞ was reduced by 55%.
Potent CYP3A4 inducers (eg, phenytoin sodium, carbamazepine, rifampin, rifabutin, rifapentine, phenobarbital, St. John’s wort [Kanjirok]) should be avoided during treatment unless no other alternative treatment options are available.
In another clinical study of pharmacokinetic interactions in healthy subjects, combination with ketoconazole, a potent CYP3A4 inhibitor, did not produce clinically meaningful effects on the pharmacokinetics of abiraterone.
Potential Effect of Abiraterone on Exposure to Other DrugsAbiraterone is an inhibitor of the hepatic drug-metabolizing enzymes CYP2D6 and CYP2C8.
In a trial examining the effect of single-dose administration of this product (plus prednisone) on the CYP2D6 substrate dextromethorphan, systemic exposure (AUC) to dextromethorphan was increased approximately 2.9-fold. The AUC of dextromethorphan, the active metabolite of dextromethorphan, was 24 increased by approximately 33%.
Caution is needed when combining this product with drugs activated or metabolized by CYP2D6 (especially drugs with a narrow therapeutic index), and lower doses of drugs with a narrow therapeutic index should be considered. Drugs that are metabolized by CYP2D6 include metoprolol, propranolol, desipramine, venlafaxine, haloperidol, risperidone, propafenone, flecainide, codeine, oxycodone, and tramadol (the latter three drugs require the formation of active analgesic metabolites via CYP2D6).
According to a trial of CYP2C8 drug-drug interactions in healthy subjects, the coadministration of pioglitazone with a single dose of 1,000 mg of this product resulted in a 46% increase in the AUC of pioglitazone and a 10% decrease in the AUC of each of the active metabolites of pioglitazone, M-III and M-IV. Although these results suggest that the increase in exposure to this product is not clinically significant when co-administered with a drug that is primarily eliminated by CYP2C8, patients should be monitored for toxic reactions caused by CYP2C8 substrates with narrow therapeutic indices when the two drugs are co-administered.
In vitro studies have shown that abiraterone sulfate and abiraterone azide, the major metabolites of this product, inhibit hepatic uptake of the transporter protein OATP1B1 and therefore may increase the concentration of drug eliminated via OATP1B1. No clinical study data are available on transporter protein-based drug interactions.
Combination with drugs known to prolongQT interval
Because depot treatment can prolong the QT interval, caution should be exercised when combining this product with drugs known to prolong the QT interval or drugs that can induce tip-twisting ventricular tachycardia, such as class IA (eg, quinidine, propyzamide) or class III (eg, amiodarone, sotalol, dofetilide, ibrit) antiarrhythmic drugs, methadone, moxifloxacin, and antipsychotics.
Co-administration with spironolactone
Spironolactone binds to androgen receptors and may increase prostate-specific antigen (PSA) levels. Combination use with this product is not recommended.
14. [Overdose]
There is limited experience with overdose of this product.
There is no specific antidote for this product. In the event of an overdose, the product should be discontinued and comprehensive supportive measures should be taken, including monitoring for arrhythmias, heart failure, and evaluation of liver function.
15. [Clinical Trials]
The efficacy and safety of this product have been demonstrated in three randomized, placebo-controlled, international clinical trials (studies COU-AA-301, COU-AA-302, and 212082PCR 3011). All patients in these studies received GnRHa or had previously undergone bilateral orchiectomy. These three trials excluded patients with prior ketoconazole treatment and a history of adrenal or pituitary disease. Because spironolactone can bind to androgen receptors resulting in elevated prostate-specific antigen (PSA) levels, the global pivotal clinical trial of this product did not allow patients to use spironolactone.
15.1. Study COU-AA-301
Patients with metastatic decompensated resistant prostate cancer previously treated with docetaxel chemotherapy
To evaluate the efficacy and safety of this product in a randomized, placebo-controlled, multicenter phase III clinical trial in patients with decompensated resistant prostate cancer previously treated with docetaxel chemotherapy. A total of 1195 patients were randomly assigned in a 2:1 ratio to receive either oral doses of 1000 mg once daily in combination with prednisone 5 mg twice daily (N = 797) or oral placebo once daily in combination with prednisone 5 mg twice daily (N = 398). Patients randomly assigned to either arm will continue treatment until disease progression (defined as a 25% increase in PSA from baseline/nadir with protocol-defined imaging progression and symptomatic or clinical progression), initiation of new antitumor therapy, intolerable toxicity, or withdrawal from the study. Patients who had previously received ketoconazole for prostate cancer and had a history of adrenal or pituitary disease were excluded from this trial.
Patient demographics and baseline disease characteristics were balanced between groups. The median patient age was 69 years (39 to 95 years), and the racial distribution was 93.3% Caucasian, 3.6% black, 1.7% Asian, and 1.6% other. 89% of enrolled patients had an Eastern Collaborative Oncology Group (ECOG) physical status score of 0 or 1, and 45% had a Brief Pain Scale score of ≥4 (most pain reported in the previous 24 hours). 90% of patients had bone metastases , 30% of patients developed visceral metastases. 70% of patients had evidence of imaging disease progression and 30% had PSA progression only. 70% of patients received one previous cytotoxic chemotherapy regimen and 30% were treated with both regimens.
A scheduled interim analysis by regimen after 552 deaths occurred showed a statistically significant improvement in overall survival (OS) for patients in the product group compared to the placebo group (Table 7 and Figure 1). After 775 deaths were observed (97% of the planned deaths in the final analysis), the survival analysis was updated. The results obtained were consistent with the results of the midterm analysis (Table 7).
Table 7: Overall survival of patients (being treated with GnRHa or previously treated with orchiectomy) treated with this product or placebo versus prednisone or prednisolone (intention-to-treat analysis set)
| Survival data analysis |
This product+Prednisone (N=797) |
Placebo+Prednisone (N=398) |
| Deaths | 333 (42%) | 219 (55%) |
| 14.8 (14.1, 15.4) | 10.9 (10.2, 12.0) | |
| < 0.0001 | ||
| Risk Ratio b (95% Confidence Interval) | 0.646 (0.543, 0.768) | |
| Updated survival data analysis | ||
| 501 (63%) | 274(69%) | |
| Median overall survival (months) (95% confidence interval) | 15.8 (14.8, 17.0) | 11.2 (10.4, 13.1) |
| 0.740 (0.638, 0.859) | ||
| a P values are based on log-rank sum tests and stratified by ECOG status score (0 or 1). b Risk ratios are based on a proportional risk ratio model after stratification. A risk ratio of <1 indicates that this product is superior. |
||
Survival rates were higher in the Benadryl-treated patients than in the placebo group at all evaluation time points within months of starting treatment (Figure 1).
Figure 1: Kaplan Meier survival curves for patients receiving this product or placebo with prednisone or prednisolone (being treated with GnRHa or prior orchiectomy combination) (Intent-to-treat analysis set)
|
| Placebo |
| This product |
| Time of death, months |
| Survival (%) |
15.2. Study COU-AA-302
Patients with metastatic depression-resistant prostate cancer without chemotherapy
Subjects enrolled in this study had not received prior chemotherapy, and subjects were asymptomatic or mildly symptomatic and had no clinical indication for chemotherapy yet. According to the Brief Pain Inventory (BPI-SF), a score of 0-1 for the most severe pain intensity in the past 24 hours was considered asymptomatic if the score was 0-1, and mildly symptomatic if the score was 2-3. Subjects with moderate or moderate pain, using opioids for cancer pain, or having visceral metastases were excluded from the study.
A total of 1088 patients were randomly assigned in a 1:1 ratio to receive either oral Benadryl 1000 mg once daily (n=546) or oral placebo once daily (n=542), and both groups were combined with prednisone 5 mg twice daily. Patients will discontinue treatment when they develop imaging or clinical (cytotoxic chemotherapy, radiotherapy or surgical treatment, opioid therapy or ECOG status score of 3 or higher) disease progression, intolerable toxicity or withdrawal from the study.
The median age of subjects treated with this product in combination with prednisone or prednisolone was 71 years, and the median age of subjects treated with placebo in combination with prednisone or prednisolone was 70 years. By ethnicity, 520 (95.4%) of the subjects in this treatment group were Caucasian, 15 (2.8%) were Black, 4 (0.7%) were Asian, and 6 (1.1%) were other. In both treatment groups, 76% of subjects had an ECOG physical status score of 0 and 24% had a score of 1. 50% of subjects had bone metastases only, 31% had bone metastases and soft tissue or lymph node metastases, and 19% had soft tissue or lymph node metastases only. The co-primary efficacy endpoints were overall survival and imaging progression-free survival (rPFS). In addition to this, the following metrics were used to assess efficacy: time to cancer pain relief with opioids, time to initiation of cytotoxic chemotherapy, time to worsening of ECOG physical status score (≥1 point compared to baseline), and time to PSA progression (as determined by the Prostate Cancer Working Group 2 [PCWG2] criteria).
Imaging progression-free survival was assessed using serial imaging examinations, as defined by PCWG2 for bone lesions and modified efficacy evaluation criteria for solid tumors (RECIST 1.1) for soft tissue lesions. rPFS was analyzed using imaging progression assessments reviewed by the central laboratory.
Based on the planned rPFS analysis, a total of 401 subjects showed imaging evidence of progression or experienced a mortality event, 150 (28%) in the Benzedrine treatment group and 251 (46%) in the placebo treatment group. There was a significant difference in rPFS between the two treatment groups (see Table 8 and Figure 2).
Table 8: Study COU-AA-302: Progression-free survival in patients treated with this product or placebo in combination with prednisone or prednisolone plus GnRHa or prior orchiectomy
| This product + Prednisone (N=546) |
Placebo+ Prednisone (N=542) |
|
| Progression-free imaging survival(rPFS) | ||
| 150 (28%) | 251(46%) | |
| Median rPFS (months) | Not met | 8.3 |
| (95% CI) | (11.66; NE) | (8.12; 8.54) |
| P-value* | <0.0001 | |
| 0.425 (0.347; 0.522) | ||
NE=not evaluated
*p values from stratified log-rank test for baseline ECOG physical status score (0 or 1)
**Risk ratio <1 makes this product more advantageous
Figure 2: Kaplan Meier curves for progression-free imaging survival in patients treated with this product or placebo in combination with prednisone or prednisolone plus GnRHa or prior orchiectomy
Subject data continued to be collected prior to a second interim analysis (IA) of OS. Imaging results for rPFS assessed by the investigator as a follow-up sensitivity analysis are shown in Table 9 and Figure 3.
Imaging progression or death occurred in 607 subjects: 271 (50%) and 336 (62%) in the Benzedrine and placebo groups, respectively. The risk of imaging progression or death was reduced by 47% in the Benadryl-treated group compared with the placebo group (HR=0.530; 95% CI: [0.451; 0.623], p<0.0001). The median rPFS was 16.5 months and 8.3 months in the product and placebo groups, respectively.
Table 9: Study COU-AA-302: Progression-free survival in patients treated with this product or placebo in combination with prednisone or prednisolone plus GnRHa or prior orchiectomy (at the time of the second mid-OS analysis-investigator review)
| This product + Prednisone (N=546) |
Placebo+ Prednisone (N=542) |
|
| Progression-free imaging survival(rPFS) | ||
| 271(50%) | 336(62%) | |
| Median rPFS (months) | 16.5 | 8.3 |
| (95% CI) | (13.80; 16.79) | (8.05; 9.43) |
| P-value* | <0.0001 | |
| 0.530 (0.451; 0.623) | ||
*p values from stratified log-rank test for baseline ECOG physical status score (0 or 1)
**Risk ratio<1, then this product is more advantageous
Figure 3: Kaplan Meier Curve for Progression-Free Imaging Survival in Patients Treated With This Product or Placebo Combined With Prednisone or Prednisolone Plus GnRHa or Prior Orchiectomy (at time of second mid-OS analysis – investigator review)
|
| This product |
| Time of death, months |
| This product |
| Placebo |
After 333 deaths were observed, a planned mid-OS analysis was performed. Based on the clinical benefit observed, this study was unblinded and subjects in the placebo group were offered this drug treatment. Compared with the placebo group, subjects in the product group had longer overall survival and a 25% lower risk of death (HR=0.752; 95% CI: [0.606; 0.934], p=0.0097), but the OS results were immature and the results of the interim analysis did not reach the statistically significant cut-off for scheduled discontinuation of the trial (see Table 10). Therefore, subjects continued to be followed for survival after this IA.
The planned final analysis of OS was performed after 741 deaths were observed (median follow-up of 49 months). Sixty-five percent (354 of 546) and 71% (387 of 542) of the subjects in the Benadryl-treated and placebo-treated groups, respectively, died. The risk of death was reduced by 19.4% in the Benadryl-treated group (HR=0.806; 95% CI: [0.697; 0.931], p=0.0033), and the OS benefit was statistically significant, with a median OS prolongation of 4.4 months (34.7 months in the Benadryl group and 30.3 months in the placebo group; see Table 10 and Figure 4). Although 44% of subjects in the placebo group received subsequent treatment with the product, there was still a significant clinical benefit advantage in the product group.
.
Table 10: Study COU-AA-302: Overall survival in patients treated with this product or placebo in combination with prednisone or prednisolone plus GnRHa or prior orchiectomy
| This product + Prednisone (N=546) |
Placebo+ Prednisone (N=542) |
|
| Interval analysis of survival | ||
| 147 (27%) | 186(34%) | |
| Median survival in months | Not reached | 27.2 |
| (95% CI) | (NE; NE) | (25.95; NE) |
| P-value* | 0.0097 | |
| 0.752 (0.606; 0.934) | ||
| Final analysis of survival | ||
| 354 (65%) | 387 (71%) | |
| Median survival in months | 34.7 | 30.3 |
| (95% CI) | (32.7; 36.8) | (28.7; 33.3) |
| P-value* | 0.0033 | |
| 0.806 (0.697; 0.931) | ||
NE=not evaluated
*p values from stratified log-rank test for baseline ECOG physical status score (0 or 1)
** Risk ratio <1 for superiority of this product
Figure 4: Kaplan Meier Curve for Survival in Patients Treated With This Product or Placebo Combined With Prednisone or Prednisolone Plus GnRHa or Prior Orchiectomy (Final Analysis)
| This product |
| Placebo |
| This product |
| Placebo |
| Time of death, months |
In addition to improved overall survival and rPFS, the product also demonstrated clinical benefit compared with placebo treatment in all of the following secondary endpoints.
Time to PSA progression determined based on PCWG2 criteria: median time to PSA progression was 11.1 months and 5.6 months for subjects in the Benadryl-treated and placebo groups, respectively (HR=0.488; 95% CI: [0.420; 0.568], p<0.0001). The time to PSA progression was approximately twice as long in the Benadryl-treated group as in the placebo group (HR=0.488). A higher proportion of subjects in the Benadryl-treated group than in the placebo group showed proven PSA remission (62% vs. 24%; p<0.0001). Among subjects with measurable soft tissue lesions, a significantly higher number of subjects in complete or partial remission was observed in the Benadryl-treated group.
Time to opioid use: At the time of final analysis, the median time to opioid use was 33.4 and 23.4 months for subjects in the Benzedrine treatment and placebo groups, respectively (HR=0.721; 95% CI: [0.614; 0.846], p<0.0001).
Time to start of cytotoxic chemotherapy: median time to start of cytotoxic chemotherapy was 25.2 months and 16.8 months for subjects in the Benzedrine and placebo groups, respectively (HR=0.580; 95% CI: [0.487; 0.691], p<0.0001).
Time to deterioration of ECOG physical status score ≥1: The time to deterioration of ECOG physical status score ≥1 was 12.3 months and 10.9 months for subjects in the Benzedrine treatment and placebo groups, respectively (HR=0.821; 95% CI: [0.714; 0.943], p=0.0053).
The following study endpoints reflect a statistically significant benefit of this treatment.
Objective remission rate: the objective remission rate was defined as the proportion of subjects with measurable lesions who achieved complete or partial remission according to RECIST criteria (baseline lymph node size needed to be ≥2 cm to be considered a target lesion). The proportions of subjects with measurable lesions at baseline who achieved objective remission were 36% and 16% in the treatment and placebo groups, respectively (p<0.0001).
Pain: The mean risk of progression in pain intensity was significantly reduced by 18% in the Benadryl-treated group compared to the placebo group (p=0.0490). The median time to progression of pain intensity was 26.7 months and 18.4 months in the Benadryl-treated and placebo groups, respectively.
Time to functional assessment of prostate cancer treatment (FACT-P) (total score) progression: The risk of FACT-P (total score) progression was 22% lower in the Benadryl-treated group compared with the placebo group (p=0.0028). The median time to worsening of FACT-P (total score) was 12.7 months and 8.3 months in the treatment group and placebo group, respectively.
15.3. Study 212082PCR 3011 (treating patients with high-risk mHSPC)
Study 212082PCR 3011 randomly assigned a total of 1199 patients with high-risk mHSPC in a 1:1 ratio to receive either this product 1000 mg once daily in combination with prednisone (5 mg once daily) (N=597) or placebo once daily (N=602). Patients enrolled in this study were newly diagnosed mHSPC patients, including those who had not received treatment prior to randomization or had been treated with ADT (orchiectomy or GnRHa with or without anti-androgens) for up to 3 months. Patients were allowed to receive 1 course of palliative radiotherapy or palliative surgery (to manage symptoms due to metastatic disease) before 1 month prior to enrollment. High-risk disease was defined as having at least two of the three risk factors at baseline: Gleason score ≥8; presence of three or more lesions on bone scan; and presence of measurable visceral metastases. Patients with significant cardiac, adrenal, or hepatic dysfunction were excluded. Patients continue to receive treatment until imaging or clinical disease progression, unacceptable toxicity, withdrawal from the study, or death occurs. Clinical progression was defined as the need for cytotoxic chemotherapy, radiation or surgery for cancer, pain requiring long-term opioid use, or worsening of the ECOG physical status score to 3 or more.
Patient demographics were balanced between groups. The median age was 67 years. The racial distribution of patients in the treatment group was 69% Caucasian, 2.5% black, 21% Asian, and 8.1% other races. 76% of patients had an ECOG physical status score of 0, 42% had a score of 1, and 3.5% had a score of 2. Based on the Brief Pain Questionnaire (most severe pain in the past 24 hours) definition, 50% of patients had a baseline pain assessment of 0 to 1 (asymptomatic), 23% had a score of 2 to 3 (mildly symptomatic), and 28% had a score of ≥4. 93.4% of patients had received prior treatment for prostate cancer, including surgery (3.8%), radiation therapy (3.8%), and endocrine therapy (93.2%) . Endocrine therapy included GnRHa or antagonists (75.0%), orchiectomy (12.0%), anti-androgen therapy (62.1%), estrogen and glucocorticoids (1.4%).
Overall survival was the primary efficacy endpoint. After 406 deaths, an interim analysis was performed as scheduled by protocol, showing that patients in the Benadryl combined with prednisone treatment group showed improved and statistically significant OS compared with the placebo group (Table 11 and Figure 5). Twenty-one percent of patients in the Benadryl combined with prednisone treatment group and 41% of patients in the placebo group received follow-up therapy that may prolong OS in metastatic CRPC, including cytotoxic chemotherapy, abiraterone acetate, enzalutamide, and systemic radiotherapy.
| Table11: Study 212082PCR 3011 Overall Survival in Patients in the Benzedrine Combination Prednisone Treatment Group and Placebo Treatment Group (Intent-to-Treat Analysis Set) | ||
| Overall Survival | This product combined with prednisone (N=597) |
Placebo (N=602) |
| Death | 169 (28.3%) | 237 (39.4%) |
| Median survival (months) (95% CI) |
NE | 34.7 (33.1, NE) |
| p-value1 | <0.0001 | |
| Risk ratio 2 (95% CI) | 0.621 (0.509; 0.756) | |
| NE=unable to estimate. 1 p values based on log-rank test and stratified by ECOG physical status score (0/1 or 2) and visceral metastases (absent or present). 2 Risk ratios were based on the stratified proportional risk model. A risk ratio of <1 indicates that this product is superior in combination with prednisone. |
||
Figure 5: Study 212082PCR 3011 Overall survival of theKaplan-Meierplot; intention-to-treat population
At the time of the planned primary analysis of rPFS, 593 events had occurred; 239 (40.0%) patients in the Benzedrine combined with prednisone treatment group and 354 (58.8%) patients in the placebo group had imaging progression or had died. A significant difference in rPFS was observed between the two treatment groups (see Table 12 and Figure 6).
| Table12: Study 212082PCR 3011 Imaging Progression-Free Survival in Patients in the Benzedrine Combination Prednisone Treatment Group and Placebo Treatment Group (Intent-to-Treat Analysis Set) | ||
| Imaging Progression-Free Survival | This product combined with prednisone (N=597) |
Placebo (N=602) |
| Disease progression or death | 239 (40.0%) | 354 (58.8%) |
| Median rPFS (months) (95% CI) |
33.0 (29.57, NE) |
14.8 (14.69, 18.27) |
| <0.0001 | ||
| Risk ratio 2 (95% CI) | 0.466 (0.394; 0.550) | |
| NE=unable to estimate. 1 P values based on log-rank test and stratified by ECOG fitness status score (0/1 or 2). 2 Risk ratios are based on the stratified proportional risk model. A risk ratio of <1 indicates superior treatment with this product plus prednisone combined with ADT. |
||
Figure 6: Study212082PCR 3011 Imaging Progression-Free Survival of Kaplan-Meier plot; intention-to-treat population
In addition to the observed benefit in overall survival and rPFS, the benefit of treatment with this product in combination with prednisone versus placebo was observed in all predefined secondary endpoints as follows.
Time to skeletal-related events: 30% reduction in the risk of skeletal-related events (HR = 0.703; 95% CI: [0.539, 0.916], p<0.0086). The median time to SRE has not been reached in both the present combined prednisone treatment and placebo groups.
Time toPSA progression (according to PCWG2 criteria): The median time to PSA progression in the Benadryl combined with prednisone treatment group was 33.2 months and 7.4 months in the placebo group (HR=0.299; 95% CI: [0.255, 0.352], p<0.0001).
Time to follow-up: The median time to follow-up had not been reached in the Benzedrine combined with prednisone treatment group at the time of the mid-period analysis and was 21.6 months in the placebo group (HR=0.415; 95% CI: [0.346, 0.497], p<0.0001).
Time to initiation of chemotherapy: The median time to initiation of chemotherapy had not been reached in the group treated with this product in combination with prednisone and was 38.9 months in the placebo group (HR=0.443; 95% CI: [0.349, 0.561], p<0.0001).
Time to pain progression: The median time to pain progression has not been reached in the Benzedrine combined with prednisone treatment group and was 16.6 months in the placebo group (HR=0.695; 95% CI: [0.583, 0.829], p<0.0001).
Most exploratory endpoints showed superiority of this product plus prednisone treatment over placebo treatment.
15.4. Clinical Trial Data in Chinese Patients (Study ABI-PRO-3002)
Patients with chemotherapy-naïve metastatic debulking-resistant prostate cancer
In a randomized, double-blind, placebo-controlled phase III clinical study of this product in combination with prednisone/prednisolone (hereafter collectively referred to as prednisone) in patients with asymptomatic or mildly symptomatic metastatic desmoid-resistant prostate cancer at 42 study centers in Asia (China, Malaysia, and Thailand) and Europe (Russia), subjects were stratified according to region (Asia or Europe) and ECOG score ( 0 or 1) were stratified and randomly assigned (1:1) to receive either this product in combination with prednisone or placebo in combination with prednisone. Eligible subjects received 1000 mg of this product (administered as 4 x 250 mg tablets) or 4 tablets of placebo (once daily) in combination with prednisone 5 mg (twice daily) on an empty stomach.
A total of 313 subjects were enrolled in this study (157 subjects: this product in combination with prednisone and 156 subjects: placebo in combination with prednisone), of which 238 Chinese patients were enrolled. Subjects received treatment until disease progression. Disease progression as defined by the study included: investigators determined that subjects experienced PSA progression (≥25% increase in PSA from nadir, ≥2ng/ml absolute increase, and confirmed after ≥3 weeks), imaging progression (confirmed progression on bone scan or progression of soft tissue disease as defined by the modified RECIST 1.1 criteria), or clinical progression (by BPI-SF assessment values ≥4 and confirmed pain progression with skeletal adverse events, increased dose of prednisone or switch to a more potent glucocorticoid, or initiation of new systemic anticancer therapy). Subjects may also discontinue treatment due to unacceptable toxicity or personal choice.
Efficacy evaluation includes disease progression as evaluated by serum PSA concentrations and assessment of survival status. The following evaluations were also performed: overall survival; objective remission rates; documentation of combined or subsequent medications; time to initiation of cytotoxic chemotherapy for metastatic prostate cancer; time to pain progression as measured by patient-reported questionnaire results and time to analgesic progression as measured by analgesic use scores; and time to clinical deterioration in ECOG physical status as determined by history review and physical examination.
Patient demographics and baseline disease characteristics were generally balanced between the two groups. The median patient age was 71 years (48 to 90 years). By the time of the protocol-prescribed interim analysis, effectiveness results showed a 58% reduction in the risk of PSA progression in subjects treated with this product in combination with prednisone compared to subjects treated with placebo in combination with prednisone (HR=0.418, p<0.0001); results for Chinese patients were consistent with the overall results, with a 58% reduction in the risk of PSA progression in subjects treated with this product compared to the placebo in combination with prednisone treatment group. subjects had a 44% lower risk of PSA progression compared to the placebo combined with prednisone treatment group (HR=0.563, p=0.0173).
Table 13: Time to PSA progression for study ABI-PRO-3002, stratified analysis (intention-to-treat analysis set)
| —————– China —————- | —————- Overall ————— | |||
| AA | Placebo | AA | Placebo | |
| Description a | (N=119) | (N=119) | (N=157) | (N=156) |
| Randomized Subjects | 119 | 119 | 157 | 156 |
| Event | 30 (25.2) | 43 (36.1) | 34 (21.7) | 60 (38.5) |
| Truncated tail | 89 (74.8) | 76 (63.9) | 123 (78.3) | 96 (61.5) |
| p-valueb | 0.0173 | <0.0001 | ||
| Risk Ratio (95% CI)c | 0.563 (0.349; 0.909) | 0.418 (0.271; 0.646) | ||
Figure 7: Kaplan Meier curves for time to PSA progression for study ABI-PRO-3002: overall (intention-to-treat analysis set)
| This product group —- Placebo group ○○○○ This product group is deleted ☆☆☆☆ Placebo group deleted |
| Random time in months |
| Country=Overall |
Figure 8: Kaplan Meier Curve for PSA Progression Time for Study ABI-PRO-3002: China (Intent-to-Treat Analysis Set)
| This product group —- Placebo group ○○○○ This product group is deleted ☆☆☆☆ Placebo group deleted |
| Placebo for this product |
| Random time in months |
| Country=Overall |
PSA remission rates were significantly higher in subjects treated with this product in combination with prednisone (67%) compared with those treated with placebo in combination with prednisone (31%) (p<0.0001). The results in Chinese patients were consistent with those of the overall population, with PSA remission rates of 67% and 37% in the Benzedrine combined with prednisone treatment group and placebo combined with prednisone treatment group, respectively (p<0.0001). Objective remission rates (complete and partial remission, CR+PR) showed a significantly higher remission rate in subjects treated with this product in combination with prednisone (23%) than in subjects treated with placebo in combination with prednisone (5%), with the former being approximately 4.8 times higher than the latter (p=0.0369). All remissions were partial remissions. The results in Chinese patients were consistent with those of the overall population, with objective remission rates of 32% and 0% in the Benzedrine combined with prednisone treatment group and placebo combined with prednisone treatment group, respectively (p=0.0052).
Safety results showed that the most frequently reported adverse events (reported by ≥10% of subjects in the drug or placebo groups) were bone pain, arthralgia, back pain, extremity pain, and hypertension. Grade 3 or 4 adverse events were reported by 17% of subjects in the product group and 21% in the placebo group; serious adverse events were reported by 4% and 7% of subjects in the two groups, respectively; adverse events leading to death were reported by 3% and 4% of subjects, respectively; and adverse events leading to discontinuation of the drug were reported by 3% and 5% of subjects, respectively.
The results of study ABI-PRO-3002 confirm a favorable clinical benefit-risk ratio for abiraterone acetate combined with prednisone treatment in mCRPC subjects who have not received chemotherapy. The safety profile of the abiraterone acetate group in this study was broadly consistent with the global pivotal phase III study COU-AA-302. No new safety signals were observed.
15.5. Clinical Trial Data in Chinese Patients (Study ABI-PRO-3001)
Patients with metastatic decompensated resistant prostate cancer previously treated with docetaxel chemotherapy
Another phase III, randomized, double-blind, placebo-controlled study of this product in combination with prednisone in patients with metastatic decompensated-resistant prostate cancer who have failed docetaxel chemotherapy in Chinese patients. All eligible subjects will be randomized in a 2:1 ratio on Day 1 of Cycle 1 to receive either 1000 mg of abiraterone acetate (administered as 4 x 250 mg tablets once daily) or placebo (4 tablets once daily) in combination with prednisone treatment (5 mg twice daily). No crossover was allowed between the two treatment groups. Each treatment cycle lasted 28 days.
A total of 214 subjects were recruited and randomized for this study. 143 subjects in the abiraterone acetate group and 71 subjects in the placebo group were included in both the ITT and safety analysis sets. Subjects received treatment until disease progression occurred. Disease progression was defined as PSA progression (Prostate Specific Antigen Working Group [PSAWG] criteria) and imaging progression (bone scan progression or soft tissue disease progression as determined by modified RECIST 1.1) with or without clinical progression (pain progression [as assessed by BPI-SF], skeletal-related adverse events, prednisone dose increase or switch to a more potent glucocorticoid , or use of additional opioid analgesics for prostate cancer-related signs and symptoms); or clinical progression with one or both of PSA progression and imaging progression.
Efficacy was assessed by measuring the subject’s PSA level during the study period. The primary efficacy endpoint was TTPP, defined as the time interval from randomization to PSA progression (as defined by PSAWG criteria). Secondary efficacy endpoints included: overall survival, PSA remission rate, objective remission rate, total quality of life score and scores on each FACT-P subscale, time to pain progression, proportion of subjects with pain relief using the BPI-SF worst pain intensity score and analgesic use score, and fatigue as assessed by the BFI.
The median age of the subjects was 68 years. Most subjects had baseline disease progression of PSA progression only. Bone metastases were observed in 95.1% and 94.4% of subjects in the abiraterone acetate and placebo groups, respectively; the proportion of subjects with pain symptoms at baseline was 72.7% and 66.2%, respectively. All subjects were medically or surgically debulked (61.7% of subjects had undergone orchiectomy and 54.7% had received GnRHa). All subjects had received chemotherapy. During the double-blind treatment period, the median treatment duration was 32.3 weeks in the abiraterone acetate group and 16.9 weeks in the placebo group. The median duration of dosing was 9 cycles in the abiraterone acetate group compared to 5 cycles in the placebo group. The median treatment duration in subjects receiving open abiraterone acetate administration during the follow-up period was 16.0 weeks, with a median dosing duration of 4 cycles (16 weeks).
The efficacy results showed a 49% reduction in the risk of PSA progression in the abiraterone acetate group compared to the placebo group (HR=0.506; p=0.0001). All subgroup analyses, except for subjects with a baseline ECOG physical status score of 2 (due to small sample size), showed that abiraterone acetate significantly improved the TTPP of subjects.
Figure 9: Kaplan Meier plot of progression time from randomization to PSA based on PSAWG criteria
(Study ABI-PRO- 3001: ITT analysis set)
| Placebo |
| This product |
| Random time in months |
In Chinese mCRPC subjects who failed previous docetaxel-based chemotherapy, treatment with abiraterone acetate in combination with prednisone significantly improved TTPP and achieved a high PSA remission rate. A clinically favorable trend in OS was also observed (HR=0.604 [0.356, 1.026]) with an HR similar to that of study COU-AA-301 (HR=0.646 [0.543, 0.768]). The rate of confirmed PSA remission was significantly higher in subjects in the abiraterone acetate group (49.7%) than in subjects in the placebo group (14.1%; relative risk=3.525; p<0.0001). A higher proportion of subjects in the placebo group (50.7%) experienced pain progression events compared to the abiraterone acetate group (37.1%). Abiraterone acetate significantly reduced the risk of pain progression by 50% compared to placebo (HR=0.496; p=0.0014). Among subjects with a pain score of 4 or higher, the rate of pain improvement was higher in the abiraterone acetate group, with a 23% difference between the two groups.
During the double-blind treatment period, 32.2% of subjects in the abiraterone acetate group reported grade 3 to 4 adverse events and 28.2% in the placebo group; 14.0% and 19.7% of subjects in the two groups reported serious adverse events during treatment; 7.0% and 9.9% of subjects reported adverse events leading to treatment discontinuation; and 6.3% and 12.7% of subjects reported adverse events leading to adverse events leading to death.
The results of study ABI-PRO-3001 confirm a favorable benefit-risk ratio for abiraterone acetate in combination with prednisone in mCRPC subjects who have received chemotherapy. The safety profile of the abiraterone acetate group in this study was broadly consistent with the global pivotal phase III study (COU-AA-301). No new safety signals were observed.
15.6. Clinical Data in Chinese Subjects in Study 212082PCR 3011
A total of 137 subjects in study 212082PCR 3011 were Chinese (this product combined with prednisone treatment group: 69 subjects; placebo group: 68 subjects). At study entry, the demographics and disease characteristics of the Chinese subjects and the overall population were generally balanced across treatment groups. The median total treatment duration was 28 months (31 cycles) and 22 months (24 cycles) for the Prednisol combined group and the placebo group, respectively. It is important to note that the results of subgroup analyses should be reviewed with caution due to the potential for chance resulting from small sample sizes.
At the time of the primary analysis of rPFS, 18 (26.1%) subjects in the Benadryl combined with prednisone treatment group and 38 (55.9%) subjects in the placebo group reported imaging progression or death. The risk of imaging progression or death was reduced by 66% in the combination prednisone group compared with the placebo group (HR = 0.341; 95% CI: 0.193, 0.605). Median rPFS had not been achieved in the Benadryl combined with prednisone treatment group and was 18.4 months in the placebo group.
At the time of the first planned mid-OS analysis, 29 subject deaths were observed: 14 (20.3%) in the Benzedrine combined with prednisone treatment group and 15 (22.1%) in the placebo group. the hazard ratio for OS was 0.862 (95% CI: 0.415, 1.788). Median survival was not achieved in either treatment group.
Subgroup analysis of secondary efficacy endpoints in Chinese subjects showed a consistent trend toward superior treatment with this product in combination with prednisone. Treatment with prednisone delayed the need for initiation of chemotherapy in Chinese subjects (HR = 0.433; 95% CI: 0.146, 1.279; median time to initiation of chemotherapy: not achieved in either treatment group), delayed the need for initiation of follow-up therapy (HR = 0.349; 95% CI: 0.173, 0.707; median time to initiation of follow-up therapy: not achieved in either treatment group), and delayed the need for initiation of follow-up therapy in Chinese subjects (HR = 0.349; 95% CI: 0.173, 0.707; median time to initiation of follow-up therapy: not achieved in either treatment group). not achieved in either treatment group), delayed pain progression in Chinese subjects (HR = 0.680; 95% CI: 0.416, 1.111; median time to pain progression: not achieved in the drug combined with prednisone treatment group and 12.9 months in the placebo group), and delayed PSA progression in Chinese subjects (HR = 0.261; 95% CI: 0.157, 0.433; median time to PSA progression time: not achieved in the Benzedrine combined with prednisone treatment group and 9.2 months in the placebo group).
Among Chinese subjects, 94.2% and 98.5% of subjects in the Benzedrine combined with prednisone treatment group and placebo group, respectively, reported treatment-emergent adverse events. The most frequently reported adverse events (reported by ≥20% of subjects in either group) were hypokalemia (37.7% and 20.6% in the combined prednisone and placebo groups, respectively), elevated ALT (30.4% and 26.5%), elevated AST (27.5% and 22.1%), hyperglycemia (24.6% and 20.6%), hypertension (24.6% and 14.7%), and dysrhythmias (24.6% and 14.7%). and 14.7%) and back pain (10.1% and 20.6%). The proportion of subjects reporting grade 3 or 4 adverse events was 60.9% and 50.0% in both groups, respectively; the proportion of subjects reporting serious adverse events was 23.2% and 32.4%, respectively; the proportion of subjects reporting adverse events leading to discontinuation was 13.0% and 16.2%, respectively; and the proportion of subjects reporting adverse events leading to a fatal outcome was 8.7%, respectively (all of which were assessed by the investigator to be non-study drug related). These events were not related to the study drug) and 1.5%.
The safety profile of this product in combination with prednisone treatment observed in Chinese subjects enrolled in Study 212082PCR 3011 was generally consistent with the overall population, and no new safety concerns were identified. Study 212082PCR 3011 demonstrated a favorable benefit-risk ratio for the use of this product in combination with prednisone in the treatment of newly diagnosed high-risk mHSPC patients in China.
16. [Pharmacologic Toxicology]
16.1. Pharmacologic effects
Abiraterone acetate is converted in vivo to abiraterone, an androgen biosynthesis inhibitor that inhibits 17α-hydroxylase/C17,20-cleavage enzyme (CYP17), which is expressed in testicular, adrenal, and prostate tumor tissues and is required for androgen biosynthesis.
CYP17 catalyzes two sequential reactions: 1) the conversion of pregnenolone and progesterone to their respective 17α-hydroxy derivatives via 17α-hydroxylase, and 2) the subsequent formation of dehydroepiandrosterone and androstenedione, respectively, catalyzed by C17,20 cleavage enzymes. Both dehydroepiandrosterone and androstenedione are androgens and are precursors of testosterone. The inhibition of CYP17 by abiraterone also leads to increased production of adrenal salt corticosteroids.
Androgen-sensitive prostate cancer can respond to androgen-lowering therapies. Androgen-blocking therapies such as GnRHa or orchiectomy reduce androgen production in the testis but do not affect androgen production in the adrenal glands or tumors.
In placebo-controlled clinical trials, abiraterone acetate caused a decrease in serum testosterone and other androgen levels in patients. There is no need to monitor the effect of this product on serum testosterone levels in clinical use.
Serum PSA levels may vary, but have not been shown to correlate with clinical benefit in individual patients.
16.2. Toxicology Studies
Repeated dosing toxicity: In repeated dosing toxicity tests in rats at 13 and 26 weeks and monkeys at 13 and 39 weeks, abiraterone acetate caused a decrease in circulating testosterone levels at an amount equivalent to approximately half the human clinical exposure (AUC). As a result, decreased organ weights and some toxicity were observed in the male and female reproductive system, adrenal glands, liver, pituitary gland (only in rats) and male mammary glands. The changes in reproductive organs were consistent with the anti-androgenic pharmacological activity of abiraterone acetate. A dose-dependent increase in the incidence of cataracts was observed in rats after 26 weeks of daily oral administration of abiraterone acetate at doses ≥50 mg/kg/day (close to the human AUC). Cataracts were not observed at higher doses (2 times higher than human AUC) in a monkey 39-week trial with daily oral administration of abiraterone acetate.
Genotoxicity: Results were negative in the abiraterone acetate and abiraterone Ames tests, the human lymphocyte cytogenetics test, and the rat micronucleus test.
Reproductive toxicity: Repeated dosing toxicity assays in male rats (13 and 26 weeks) and monkeys (39 weeks) at doses of ≥50 mg/kg/day (rats) and ≥250 mg/kg/day (monkeys) showed reproductive atrophy, azoospermia/seminopenia, and proliferative changes with effects consistent with the anti-androgenic pharmacological activity of abiraterone. AUCs for these effects were observed in rats and monkeys that were close to and approximately 0.6 times the clinical exposure in humans, respectively.
In the rat fertility and early embryonic development toxicity assay, reduced reproductive system organ weights, reduced sperm counts, reduced sperm viability, altered sperm morphology and reduced fertility were seen in male rats given 30 mg/kg/day and higher doses for 4 weeks. Mating of unadministered female rats with male rats given the 30 mg/kg/day dose resulted in a decrease in the number of corpus luteum, a decrease in the number of embryos that were born and survived, and an increase in the rate of loss before implantation. The effects of abiraterone acetate on fertility in male rats recovered after 16 weeks of drug withdrawal. Administration of abiraterone acetate at 30 mg/kg/day and higher to female rats from two weeks prior to mating to day 7 of gestation caused an increased incidence of irregular or delayed estrous cycles and increased rates of pre-terminal loss (300 mg/kg/day). No differences were observed in various parameters of mating ability, fertility and their offspring in female rats administered abiraterone acetate. The effects of abiraterone acetate on female rats recovered after 4 weeks of drug withdrawal. Converted to body surface area, the rat dose of 30 mg/kg/day is approximately 0.3 times the recommended human dose (1000 mg/day).
In a rat embryo/fetus developmental toxicity assay, oral administration of abiraterone acetate at 10, 30 and 100 mg/kg/day (approximately 0.03, 0.1 and 0.3 times the human AUC, respectively) on days 6-17 of gestation caused developmental toxicity, with embryo/fetus mortality (increased post-arrival loss and fetal resorption rates, decreased number of live fetuses) seen at ≥10 mg/kg/dose. The dose of ≥30mg/kg/dose may cause shortening of anal genital distance of fetus, and 100mg/kg/dose may cause low weight of fetus. A dose of ≥10mg/kg/dose may cause maternal toxicity.
Carcinogenicity: A two-year carcinogenicity test in rats administered orally showed that abiraterone acetate at doses of 5, 15, and 50 mg/kg/day in male rats and 15, 50, and 150 mg/kg/day in female rats caused testicular mesenchymal cell adenoma and mesenchymal cell carcinoma at all doses, thought to be related to the pharmacological activity of abiraterone. Abiraterone acetate was not seen to be carcinogenic in female mice at 0.8 times the human exposure. No carcinogenicity was seen in a 6-month carcinogenicity test in Tg.rasH2 transgenic mice.
17. [Pharmacokinetics]
The pharmacokinetics of this product and its active metabolite abiraterone have been studied in healthy subjects and patients with mCRPC. In vivo, the product is rapidly converted to abiraterone. In clinical studies, >99% of the analyzed samples had plasma concentrations of this product below detection levels (<0.2 ng/ml).
17.1. Absorption
The median time to peak of abiraterone was 2 hours after oral administration of this product in patients with mCRPC. Steady-state accumulation of abiraterone was observed with an exposure (steady-state AUC) twice that of a single dose of 1000 mg of this product.
In patients with mCRPC, Cmax and AUC steady-state values (mean±SD) were 226±178 ng/ml and 993±639 ng∙h/ml, respectively, at 1000 mg once-daily doses. no major deviations from dose proportionality were observed in the dose range 250-1000 mg. There was no significant increase in exposure when the dose was increased from 1000 mg to 2000 mg (mean AUC increased by 8%).
Systemic exposure to abiraterone was elevated when this product was administered with food. Abiraterone Cmax and AUC0-∞ increased to approximately 7-fold and 5-fold, respectively, when this product was taken concomitantly with a low-fat meal (7% fat, 300 calories); these values increased to approximately 17-fold and 10-fold, respectively, when this product was taken concomitantly with a high-fat meal (57% fat, 825 calories). Given the diversity and variability of foods, concomitant administration of this product with food may result in elevated and variable exposure. Therefore, food should not be consumed for at least 2 hours prior to and 1 hour after administration. In addition, this product must be taken in whole tablets with water (see [DOSAGE]).
17.2. Distribution and protein binding
Abiraterone is highly bound (>99%) to human plasma proteins, albumin, and alpha-1 acidic glycoprotein. The steady-state apparent volume of distribution (mean ± SD) was 19,669 ± 13,358 L. In vitro studies showed that neither this product nor abiraterone was a substrate for P-glycoprotein at clinically relevant concentration ranges, and that this product was an inhibitor of P-glycoprotein.
17.3. Metabolism
Following oral administration of 14C-abiraterone acetate capsules, abiraterone acetate is hydrolyzed to abiraterone (the active metabolite). This process may be converted by the action of esterases (esterases have not been identified) rather than being mediated by CYP. The two major circulating metabolites of abiraterone in human plasma are abiraterone sulfate (inactive) and N-oxidized abiraterone sulfate (inactive), each accounting for approximately 43% of the exposure.CYP3A4 and SULT2A1 are involved in N-oxidized abiraterone sulfate formation, and SULT2A1 is also involved in abiraterone sulfate formation.
17.4. Excretion
In patients with mCRPC, the mean terminal half-life (mean ± SD) of abiraterone in plasma was 12 ± 5 hours. After oral administration of 14C-abiraterone acetate, approximately 88% and 5% of the radioactive dose was recovered from feces and urine, respectively. The major compounds present in the feces were the prototype of the product and abiraterone (55% and 22% of the administered dose, respectively).
17.5. Patients with hepatic impairment
The pharmacokinetics of abiraterone were evaluated in subjects with mild (n = 8) or moderate (n = 8) hepatic impairment (Child-Pugh class A and B, respectively) at baseline and in 8 healthy subjects with normal liver function. Systemic exposure to abiraterone increased approximately 1.1-fold and 3.6-fold following a single oral dose of 1000 mg on an empty stomach in subjects with mild and moderate hepatic impairment at baseline, respectively. The mean half-life of abiraterone was prolonged to 18 and 19 hours in subjects with mild and moderate hepatic impairment, respectively.
Another trial analyzed the pharmacokinetics of abiraterone in eight subjects with severe hepatic impairment (Child-Pugh class C) at baseline and eight healthy subjects with normal liver function. Subjects with severe hepatic impairment at baseline had an approximately 7-fold increase in systemic exposure (AUC) to abiraterone compared to subjects with normal hepatic function. In addition, the trial found that the mean protein binding rate was lower in subjects with severe hepatic impairment at baseline than in subjects with normal hepatic function, resulting in a 2-fold increase in exposure to the free drug fraction in subjects with severe hepatic impairment. (See [DOSAGE])
17.6. Patients with renal impairment
The pharmacokinetics of abiraterone were evaluated in patients with end-stage renal disease (n=8) and in subjects with normal renal function (n=8) receiving a stable hemodialysis regimen. In the group of patients with end-stage renal disease, a single oral dose of 1000 mg of this product was administered 1 hour after dialysis on an empty stomach and sampled for pharmacokinetic analysis within 96 hours of dosing. Results showed that systemic exposure to abiraterone was not elevated after a single oral dose of 1000 mg in subjects with end-stage renal disease receiving dialysis compared with subjects with normal renal function (see [Precautions]).
18. [Storage]
Store between 15 and 30°C.
19. [Packaging]
High-density polyethylene round bottle, 120 tablets/bottle
20. [Expiration date]
24 months.
21. [Executive Standard]
JX20130141
22. [Imported drug registration certificate number]
H20150264
23. [Manufacturer]
Company name: Patheon Inc.
Production Address: 2100 Syntex Court, Mississauga, Ontario, L5N 7K9, Canada
23.1. Domestic Contact Information
Name: Xi’an Janssen Pharmaceutical Co.
Address: No. 19, Cao Tang Road 4, Cao Tang Science and Technology Industrial Base, Xi’an High-tech Zone
Zip code: 710304
Inquiry number: 400 888 9988
Fax number: (029) 82576616
Website :http://www.xian-janssen.com.cn