Treatment Guidelines for Renal Cell Carcinoma
(2022 Edition)
I. Overview
Renal cell carcinoma (Renal cell carcinoma, RCC) is a malignant tumor originating from the renal tubular epithelium, accounting for 80% of renal malignancies to 90%. The most common histopathological type of renal cell carcinoma is clear cell carcinoma, followed by papillary renal cell carcinoma and suspicious cell carcinoma, as well as rare types of renal cell carcinoma such as collecting duct carcinoma. With the development of medical imaging, the detection rate of early renal cell carcinoma is gradually increasing.
Longer, limited renal cell carcinoma is detected after nephrectomy with preservation of the renal unit or radical nephrectomy (radical nephrectomy). “font-family:Times New Roman”>radical nephrectomy, RN ) can achieve a satisfactory outcome. According to statistics, the number of patients with advanced disease at diagnosis has decreased from 30% a few years ago to 17%, and with the continued development of targeted therapy continues to develop and the rise of immunotherapy, the outcome of advanced renal cell carcinoma has gradually improved.
II. Epidemiology and etiology
(A) Epidemiology.
Worldwide, the incidence of renal cell carcinoma accounts for approximately 2% of adult malignancies to 3% of adult malignancies, with significant geographical differences in distribution, with the highest incidence in developed Western countries such as North America and Western Europe and the lowest incidence in developing countries such as Africa and Asia. According to GLOBOCAN2020 global cancer statistics, by 2020 the global incidence of renal cell carcinoma is the incidence is the 14th most common malignancy, lower than that of the urinary tract
prostate cancer and bladder cancer, with the 15th mortality rate. The age-standardized incidence of renal cell carcinoma was 6.1/100 million in men and 3.2/100 million in women. Age-standardized
The mortality rate in men was 4.6/10 million for men and 1.8/10 million for women. According to the 2018 Chinese Tumor Registry Annual Report, the number of new cases of renal cell carcinoma in China in 2015 accounted for 17th of malignant tumors and the 18th of deaths. The crude incidence rate of renal cell carcinoma in China is 4.02/100 million, with an age-standardized incidence rate of 2.66/100 million. The crude incidence rate of renal cell carcinoma in men was 5.10/10 million and the age-standardized incidence rate was 3.43/10 million The incidence of renal cell carcinoma in women was 2.92/100 million with an age-standardized incidence of 1.89/10 million. million.
(ii) Etiology.
The etiology of renal cell carcinoma is unclear, and its development has been associated with genetics, smoking, and obesity.
1 Hereditary factors
Most renal cell carcinomas are sporadic, and hereditary renal cell carcinomas account for 2% of all renal cell carcinomas~. span style=”font-family:Times New Roman”>4%, are mostly inherited in families in an autosomal dominant manner and are caused by different genetic variants, which include both oncogenes and oncogenes. Well-defined hereditary renal cell carcinomas include Hippel– Lindau
(von Hippel-Lindau, VHL) disease (bilateral multiple renal clear cell carcinoma and renal cysts), MET gene-related hereditary papillary renal cell carcinoma, abnormalities of the yohimbe acid hydratase gene caused by hereditary smooth muscle tumor disease and renal cell carcinoma, Burt–Hogg–Dubb (Birt-Hogg-Dube, BHD) syndrome (multiple renal suspicious cell carcinoma, heterogeneous suspicious cell and eosinophilic renal tumors, papillary renal cell carcinoma), HRPT2 gene-related hyperparathyroidism –mandibular tumor syndrome ( mixed epithelial and stromal tumors, papillary renal cell carcinoma) (Table 1). The following groups are generally considered to be potential patients for hereditary renal cell carcinoma: 1)
≤45 years of age; (ii) bilateral / multiple renal tumors; (iii) family history of renal cell carcinoma (at least 1 first-degree relatives and at least 2 second-degree relatives); ④ history of renal cell carcinoma combined with other tumors (pheochromocytoma, gastrointestinal mesenchymal tumor, neurological hemangioblastoma, pancreatic neuroendocrine tumor, etc.), combined with other lesions such as pulmonary cysts, spontaneous pneumothorax, etc.; ⑤ Combination of rare skin lesions (smooth muscle sarcoma, angiofibroma, etc.); 6) Personal or family history of renal cell carcinoma-related syndrome. For this group of patients, genetic mutation testing may be recommended for the individual and their family members.

Table 1 Common hereditary renal cell carcinomas and clinical manifestations
|
Syndrome Abbreviation |
Mutant loci |
Pathology Type |
Clinical presentation |
||
VHL |
VHL |
ccRCC |
ccRCC, pheochromocytoma, pancreatic kidney |
||
|
Organic cysts, neurological retinal vessels |
|||||
|
Blastoma, paraganglioma, pancreatic |
|||||
|
Endocrine tumors, lymphoid cystic tumors, ependymal< |
|||||
|
Testicular adenoma |
|||||
|
HPRC |
MET |
pRCC Ⅰ |
pRCC |
||
|
BHD |
FLCN |
FLCN |
Multiple RCC |
Susceptible cell carcinoma, mixed eosinophilic |
|
|
Tumor, Fibroblastoma, Dermatome, Pulmonary Cyst |
|||||
|
swelling, pneumothorax |
|||||
|
HLRCC |
FH |
< span style="font-size:12pt">pRCC II |
pRCC, cutaneous uterine smooth muscle tumor, |
||
|
Uterine Smooth Muscle Sarcoma |
|||||
|
SDH RCC |
SDHB,SDHD, .
SDHC |
ccRCC, the
chromophob |
ccRCC, suspicious cells, eosinophilic
Pheochromocytoma, paraganglioma |
e RCC
cowden syndrome
PTEN
ccRCC
ccRCC, breast cancer, follicular thyroid cancer, endometrial cancer MITF Related tumors MITFRCC melanoma, PECOMAHPT-JTHRPT2 nephroblastoma multiple RCC, nephroblastoma, hyperparathyroidism, thyroid cancer BAP1 Related tumors BAP1 ccRCC ccRCC, uveal melanoma, melanoma, mesothelioma Chromosomal translocations [t(3;8), t
(2;6)] related tumors FHIT/FRA3B on
chr3, RNF139 on chr8ccRCCccRCC, papillary thyroid carcinoma
Note: VHL, Hippel-Lindau disease; ccRCC, clear cell renal cell carcinoma; HPRC, hereditary papillary renal carcinoma; pRCC, papillary renal cell carcinoma; BHD, Burt-Hogg-Dubb syndrome; HLRCC, hereditary smooth muscle disease and renal cell carcinoma; HPT-JT, hyperparathyroidism-jaw tumor.
Smoking
Smoking can increase the risk of renal cell carcinoma, and prospective studies have concluded that smoking is a moderate risk factor. The relative risk of renal cell carcinoma in individuals with a previous history of smoking was 1.3, while the relative risk in individuals who were smoking was 1.6. 1.6.
- style=”margin-left: 79pt”>
- Obesity
The degree of obesity is generally expressed as body mass index, and an increase in body mass index is associated with an increased risk of renal cell carcinoma. The specific ways in which obesity increases the risk of renal cell carcinoma
The mechanism is unknown and may be related to increased androgen and estrogen release from obesity or to the release of some cytokines from adipocytes.
- style=”margin-left: 79pt”>
- Acquired renal cysts associated with long-term dialysis in end-stage renal disease
Patients with end-stage renal disease have a higher incidence of renal cell carcinoma compared to the general population. Patients on long-term dialysis are at risk for acquired renal cysts. In these patients with renal cell carcinoma, the tumors are usually bilateral, multiple, and histologically papillary.
- style=”margin-left: 79pt”>
- Other
There is evidence that alcohol consumption, occupational exposure to trichloroethylene, and women with high estrogen levels may increase the risk of renal cell carcinoma. Further research is needed to investigate the potential impact of the interaction between genetic factors and environmental exposures.
III.
(I) Gross pathology.
Most renal cell carcinomas occur in one kidney, and bilateral renal cell carcinomas (heterozygous or simultaneous) account for only 2% of sporadic renal cell carcinomas to 4%. Renal tumors are often solitary, with 10% to 20% being multifocal. Multifocal cases are commonly seen in patients with hereditary renal cell carcinoma as well as papillary renal cell carcinoma. The tumors vary widely in size and often have a pseudo-envelope separating them from the surrounding renal tissue.
(ii) Classification.
1981, 1997, 1997, 1997, 1997. span>year, 2004 and 2016 year family:Times New Roman”>WHO has launched a total of 4
versions of the classification criteria for renal tumors. The current clinical use is 2016 year
WHO version 4 of the The classification criteria for renal tumors (Table 2), which follows the 2004 edition
The framework of this publication has been updated to include only some of the more recognized tumors, such as updating multifocal cystic renal cell carcinoma, which has never been reported as a recurrent metastasis, to low-grade malignant potential multifocal cystic renal cell tumor, and updating < span style="font-family:Times New Roman">Xp11.2 translocation
/TFE3 fusion gene-associated renal cell carcinoma is classified as MiT family translocated renal cell carcinoma, which also includes TFEB gene translocated renal cell carcinoma, among others. In addition, some new pathological subtypes have been added. As mentioned earlier, renal cell carcinoma can be divided into hereditary renal cell carcinoma and sporadic renal cell carcinoma based on the relationship with genetic syndromes. The pathologic pattern alone cannot distinguish hereditary renal cell carcinoma from sporadic renal cell carcinoma.
 < h2>Table 2 WHO pathologic histologic classification of renal cell tumors, 2016
< h2>Table 2 WHO pathologic histologic classification of renal cell tumors, 2016
Renal cell tumors Renal cell tumors


Clear cell renal cell carcinoma Clear cell renal cell carcinoma
Low malignant potential multifocal cystic renal cell carcinoma Multilocular cystic renal neoplasm of low malignant potential
Papillary renal cell carcinoma Papillary renal cell carcinoma
Hereditary smooth muscle tumor disease and renal cell carcinoma-associated renal cell carcinoma
Hereditary leiomyomatosis and renal cell carcinoma (HLRCC)-associated renal cell carcinoma
Smoldering renal cell carcinoma Chromophobe renal cell carcinoma
Collecting duct carcinoma Collecting duct carcinoma
Renal medullary carcinoma Renal medullary carcinoma
MiT Familial translocated renal cell carcinoma MiT Family translocation carcinomas
Succinate dehydrogenase deficient renal cell carcinoma Succinate dehydrogenase (SDH)-deficient renal carcinoma
Mucinous tubular and spindle cell carcinoma Mucinous tubular and spindle cell carcinoma
Tubular cystic renal cell carcinoma Tubulocystic renal cell carcinoma
Acquired cystic renal cell carcinoma Acquired cystic disease associated renal cell carcinoma
clear cell papillary renal cell carcinoma Clear cell papillary renal cell carcinoma
Uncategorized renal cell carcinoma Renal cell carcinoma, unclassified
Papillary adenoma Papillary adenoma
 < span style="font-size:12pt">Eosinophilic adenoma Oncocytoma
< span style="font-size:12pt">Eosinophilic adenoma Oncocytoma
- style=”margin-left: 79pt”>
- Characteristics of common renal cell carcinoma pathology
-
Clear cell renal cell carcinoma: Clear cell renal cell carcinoma is the most common pathologic subtype of renal cell carcinoma, accounting for approximately 1,000% of renal cell carcinomas. span>60%~85%.
- style=”margin-left: 55pt”>
- Gross examination: equal bilateral renal prevalence, less than 5%of cases can
occur multicentrically or involve bilateral kidneys. It appears as a solid round nodule in the renal cortex, well-defined or indistinct from the surrounding renal tissue, with pseudo-envelope; the tumor may appear colorful or golden yellow on the cut surface, with common necrosis, hemorrhage, cystic changes, and occasional calcification or ossification on the cut surface.
- Gross examination: equal bilateral renal prevalence, less than 5%of cases can
- Histopathology: clear or eosinophilic cytoplasm of cancer cells with clear cytosol; small thin-walled blood vessels forming a ciliated vascular network; nested and glandular vesicle-like structures of tumor cells; tumor giant cells can be seen in sarcomatoid differentiated tumor areas. The tumor cells in the sarcomatoid differentiation can be seen as tumor giant cells, and those in the rhabdoid differentiation can be seen as wide eosinophilic cytoplasm with eccentric nuclei and prominent nucleoli, suggesting poor prognosis; necrosis, fibrous mucus-like interstitium and calcification and ossification can be seen in some tumors.
- Commonly used immunohistochemical antibodies:Pax-8,CA9,MUC1,MUC3, CK8, ,CK18,,,vimentin,CD10< span style="font-family:Arial">and EMA positive. Immunohistochemistry
Staining is not necessary for diagnosis and is used only in difficult cases.
-
Papillary renal cell carcinoma: Papillary renal cell carcinoma accounts for approximately >7%~14% , is the second most common renal cell carcinoma after clear cell renal cell carcinoma. Its age of onset, male-to-female incidence ratio, and symptoms and signs are similar to those of renal clear cell carcinoma.
- Grand body examination: lesions involving bilateral kidneys and multifocal are more common than clear cell carcinoma; they are mostly grayish-pink in color, and hemorrhage, necrosis, and cystic changes are common.
- Histopathology: the lesions are classified into type I and type II based on histopathological changes 2 subtypes. The tumor cells consist of papillary or tubular structures with a ciliated vascular axis, with foamy macrophages and cholesterol crystals in the papillary core; the tumor cells are small, with sparse cytoplasm (type I) or with abundant eosinophilic cytoplasm and high nuclear grade (type II), with areas of necrosis, sarcomatoid differentiation, and rhabdomyolysis. The prognosis of patients with type I papillary renal cell carcinoma is better than that of patients with type II.
-
Commonly used immunohistochemical antibodies: Similar to clear cell renal cell carcinoma, existing studies have shown that the prognosis of patients with type I papillary renal cell carcinoma is better than that of patients with type II. cell carcinoma, the available studies suggest that papillary renal cell carcinoma CK7 is positive, “font-family:Times New Roman”>P504S positive, and type I was more positive than type II. Immunohistochemical staining is not necessary for diagnosis and is used only in difficult cases.
- Suspected cell carcinoma: Suspected cell carcinoma accounts for approximately 4%~10% 10% “font-family:Arial”>. It can develop from childhood to old age, with an average age of 60 years and an approximately equal incidence in both sexes. There are no specific clinical signs and symptoms compared to other renal cell carcinoma subtypes. The tumor is large and uniform in density or signal, without hemorrhage, necrosis, or calcification.
- Major examination: the tumor is unencapsulated but well-defined, with a uniform brownish texture on the cut surface, and necrosis is seen, but hemorrhagic foci are rare.
-
Histopathology: The tumor is solid and lamellar, but may also be small nests, microcysts, beams, and occasional papillae. Unlike clear cell carcinoma, the blood vessels in the tumor are thick-walled rather than thin-walled; the tumor cells are large, polygonal, with a clear, slightly reticulated cytoplasm and very clear cell membranes (suspicious cells), or eosinophilic cytoplasm. The histologic features include: small cells with a slender eosinophilic granular cytoplasm; eosinophilic cells located in the center of the cell nest and pale cytoplasmic cells located at the periphery of the cell nest; irregularly crinkled nuclei with clumped chromatin, binucleated cells and perinuclear haloes; and round cells.
- style=”margin-left: 55pt”>
- Commonly used immunohistochemical antibodies:CK,CK7 ,EMA,< span style="font-family:Times New Roman">lectinsand
parvalbumin positive, weakly positive for renal cell carcinoma antigen, vimentin and CD10 negative. Immunohistochemical staining is not necessary for diagnosis and should be used only in difficult cases.
- style=”margin-left: 55pt”>
-
Special staining Hale Colloidal iron shows diffuse positivity of tumor cells.
- Commonly used immunohistochemical antibodies:CK,CK7 ,EMA,< span style="font-family:Times New Roman">lectinsand
-
Low malignant potential multiatrial cystic renal tumor: In the 2016 pre-2016 version, this tumor was known as multiatrial cystic renal cell carcinoma. The tumor consisted of multihoused cysts with walls containing a single layer or clusters of distributed clear cells without an expansile growth pattern; the morphology was indistinguishable from clear cell carcinoma (G1/2) cannot be distinguished without necrosis, vascular invasion, and sarcomatoid differentiation. Needs to be distinguished from renal clear cell carcinoma with cystic changes, extensive vitreous
degeneration, hemorrhage, and iron-containing hemoglobin deposition. The immunophenotype is similar to that of clear cell renal cell carcinoma.
-
Collecting ductal carcinoma and renal medullary carcinoma: renal collecting ductal carcinoma is a malignant epithelial tumor arising from the Bellini’s collecting duct; renal medullary carcinoma The patients almost always have sickle cell hematologic disease. The two have some similarities in their gross and histologic manifestations and are described together.
- Gross examination: both occur in the central part of the kidney, in the medulla, with solid, gray-white, indistinct borders and visible necrosis.
-
Histopathology: It should be noted that Bellini’s collecting duct carcinoma is often a diagnosis of exclusion, and the site of the tumor is important in making the diagnosis. Histologically, irregular tubular structures with highly heterogeneous cells are seen; microscopically, the tumor of renal medullary carcinoma is hypodifferentiated and lamellar in distribution, with adenoid cystic arrangement of tumor cells and more neutrophil infiltration within the tumor, along with sickle-shaped red blood cells.
-
Commonly used immunohistochemical antibodies: The two common immunohistochemical combinations include PAX2, PAX8< span style="font-family:Arial">,OCT3/4,< span style="font-family:Times New Roman">SMARCB1/INI1,, P63.
- style=”margin-left: 48pt”>
- Pathologic features of rare renal cell carcinoma types
- Pathologic features of rare renal cell carcinoma types
-
MiT family transposition carcinoma: two types of tumors are included, each associated with two transcription factors (TFE3 and TFEB) appear to be fusion gene related. Xp11 translocation causes TFE3 gene fusion;t(6;;11) caused by MALAT1-TFEB< span style="font-family:Arial">fusion. This tumor is common in childhood and accounts for only 1.6%to 4%. t(6;11) renal cell carcinoma is less common than Xp11transposed renal cell carcinoma. It is largely non-characteristic. Microscopically,Xp11 translocated renal cell carcinoma shows papillae formed by clear cells with scattered grit-like calcifications; whereas >t(6) span>;11) translocation
Sexual renal cell carcinoma presents as a nest of cells composed of two types of cells, large and small. Basement membrane-like material was seen deposited within them. Immunohistochemistry showed decreased expression of epithelial markers, such as CK, EMA; expression of PAX8 and other renal tubular markers; Xp11 renal cell carcinoma: partial expression of melanin markers and TFE3; t(6;11 ) renal cell carcinoma constant expression of melanin markers such as HMB45, Melan A, and Cathepsin K, TFEB. FISH detects TFE3 or TFEB fusion genes.
- Acquired cyst-associated renal cell carcinoma: These tumors often have a history of end-stage renal disease and acquired cystic kidney disease, most commonly in patients on long-term hemodialysis. They are usually bilateral, multifocal lesions with well-defined borders and a background of surrounding polycystic kidneys. The histology is characterized by microcystic structures and abundant eosinophilic oxalate crystals within the tumor. Immunohistochemistry showed renal cell carcinoma, CD10 and and CD10. family:Times New Roman”>AMACR positive,CK7 negative.
-
Clear cell papillary renal cell carcinoma: These tumors account for 1%to4%of renal tumors. Roman”>4%, with no gender predilection, disseminated or associated with end-stage renal disease, VHL syndrome. The gross presentation is a small, well-defined, enveloped mass, often with cystic changes. The histologic appearance is papillary with uniform cell size, hyaline cytoplasm, nuclei arranged away from the basement membrane, and visible cytoplasmic protrusions,G1 or. span>G2 cell nuclear grading, tumor necrosis, extra-renal invasion, and vascular tumor emboli were rare. Immunohistochemistry showedCK7 diffusely positive,CAIX cuprate positive,PAX2,PAX8and CK34βE12 E12E12 span>positive,P504S andpositive,and< CD10 negative. It should be noted that in cases of atypical morphology: if P504S orCD10 positive,CK7 < span style="font-family:Arial">Diffuse positive attenuated, better diagnosed as clear cell carcinoma.
- style=”margin-left: 72pt”>
- Hereditary smooth muscle tumor disease and renal cell carcinoma-associated renal cell carcinoma.
- Hereditary smooth muscle tumor disease and renal cell carcinoma-associated renal cell carcinoma.
Hereditary smooth muscle tumor disease and renal cell carcinoma-associated renal cell carcinoma is a group of renal cell carcinomas with germline mutations in the gene for johumate hydratase, associated with extra-renal smooth muscle tumor disease. The tumor may be grossly cystic in appearance, with marked attachment nodules.
Histologic morphology is similar to that of papillary renal cell carcinoma or to that of collecting duct carcinoma; cells
The nucleus showed intranuclear inclusion bodies and a perinuclear halo. Immunohistochemistry showed absent expression of ferredoxin hydratase.
- Succinic acid dehydrogenase-deficient renal cell carcinoma: These tumors are rare and are mostly hereditary. The tumor is a solid mass with well-defined borders. The tumor cells are arranged in solid, nested or tubular structures; the cytoplasm is vacuolated and eosinophilic to hyaline, the nuclear contours are regular and smooth, the chromatin is fine, and the nucleoli are inconspicuous (similar to neuroendocrine cells); the cytoplasm is vacuolated; occasionally, high-grade nuclei are seen. Immunohistochemistry showed a deficiency in succinate dehydrogenase expression, with succinate dehydrogenase B expression being the most common deficiency.
- Tubular cystic carcinoma: These tumors are rare and are often incidental on physical examination. The bulk has a grayish spongy or Swiss cheese-like appearance. Microscopically, they appear as small to medium-sized tubes with large cystic lumen formation, lined with a single layer of flat, cuboidal, or columnar epithelium, with spike-like cells; the nuclei are equivalent to G3of the nucleus. Immunohistochemistry expresses high molecular weight keratin.
-
Mucinous tubular and spindle cell carcinomas: these tumors Less than 1% of renal tumors. A largely solid mass with well-defined borders is present. Histology showed elongated or interlocking tubular structures with some areas of spindle cells; low-grade nuclei; and basophilic mucinous interstitium. Immunohistochemistry showed CK7, PAX2andP504S
positive.
-
Renal cell carcinoma, unClassification:Currently includes carcinomas that do not have the characteristics of existing renal cell carcinoma subtypes and can be low-grade or high-grade. The following types are included: renal cell carcinomas containing 1 or more pathologic features of renal cell carcinoma, renal cell carcinoma with mucus secretion, renal cell carcinoma with unclassified epithelial component, renal cell carcinoma, low-grade or high-grade unclassified eosinophilic tumors, and sarcomatoid carcinoma. This classification will become less common as more is known about renal cell carcinoma.
(iii) Classification.
Pathologic grading is an important prognostic correlate that applies only to clear cell renal cell carcinoma and papillary renal cell carcinoma. In previous versions of the WHO classification, the most commonly used is the 1982 Fuhrman 4 classification. The 1998 WHO recommended the Fuhrman grades I and II into a single grade, i.e., highly differentiated, grade III as moderately differentiated, and grade IV as hypodifferentiated or undifferentiated. The 2016 version of the pathology grading has been further adjusted from the original Fuhrman four-grade grading system by adding objective The evaluation criteria were added to form the WHO/ISUP pathology grading system (Table 3), making it more operational and Repeatability is better.
Table 3 WHO/ISUP Nuclear Classification Criteria for Renal Cell Carcinoma, 2016 Edition
Grading |
Definition |
1 level |
Nuclei are absent or inconspicuous under 400× microscopy and are basophilic |
2 levels |
Nuclei are obvious under 400× microscopy, eosinophilic, visible but not prominent under 100× microscopy |
|
3 levels |
100× nuclei clearly visible, eosinophilic |
4 levels |
Significant nuclear pleomorphism, multinucleated tumor giant cells and/or rhabdoid and/or sarcomatoid differentiation are seen |
(iv) Staging.
The most widely used staging for renal cell carcinoma is the American Joint Committee on Cancer Staging (American Joint Committee on Cancer Staging (AJCC) has developed the TNM staging system. span>staging system, currently applied in the 2017 update of version 8 . See Table 4 and Table 5 for details.
 < img src="https://www.kiraspecialist.com/wp-content/uploads/2022/06/062222_1307_20227.png" alt=""/>
< img src="https://www.kiraspecialist.com/wp-content/uploads/2022/06/062222_1307_20227.png" alt=""/>
Table 4 2017 8th Edition AJCC TNM Staging for Renal Cell Carcinoma
Staging Standard primary tumor (T)
|
TX |
Primary Tumor Unable to evaluate |
|
|
T0 |
No evidence of primary tumor |
|
|
T1 |
Tumor with a maximum diameter ≤ 7cm and confined to the kidney |
|
|
T1a |
Tumor with a maximum diameter ≤ 4cm and confined to the kidney |
|
|
T1b |
4cm |
|
|
T2 |
Massive tumor >7cm in diameter and confined to the kidney |
|
|
T2a |
7cm < maximum tumor diameter ≤10cm and confined to the kidney |
|
|
T2b |
Tumor is confined to the kidney, with a maximum diameter >10cm and confined to the kidney |


|
T3 |
Tumor invaded major vein or perinephric tissue but did not invade the ipsilateral adrenal gland and was not super |
|||
|
over perirenal fascia |
||||
|
T3a |
Tumor invades the renal vein or its branching renal segmental veins, or invades the renal pelvic system, or invades |
|||
|
Committed perirenal fat and/or sinus fat, but not beyond perirenal fascia |
||||
|
T3b |
Tumor invades the subphrenic vena cava |
|||
|
T3c |
||||
|
T4 |
T4 |
Tumor invades the perirenal fascia, including the ipsilateral adrenal gland that invades the adjacent tumor |

NX Regional lymph nodes could not be evaluated
N0 regional lymph nodes without metastasis
N1 Regional lymph nodes with metastasis

M0 No distant transfer
 < span style="font-size:12pt">M1 with distant transfer
< span style="font-size:12pt">M1 with distant transfer

Table 5 Clinical staging/prognostic subgroups for renal cell carcinoma
Stage Tumor status


I Phase T1 N0 M0
Phase II T2 N0 M0
Phase III T1/2 N0 M0
T3 N0/1 M0
IV Phase T4 Any N M0
 < span style="font-size:12pt">Any T Any N M1
< span style="font-size:12pt">Any T Any N M1
IV. Diagnosis
(A) Clinical manifestations.
The clinical manifestations of patients with renal cell carcinoma are complex and variable. Some of these clinical manifestations are directly caused by the renal tumor itself, while others may be due to hormones secreted by the cancer cells or metastases. The majority of patients who present to the hospital with renal cell carcinoma are usually detected inadvertently by imaging because of the increasing popularity of health screening.
In clinical practice, early-stage renal cell carcinoma often lacks clinical presentation. When the classic
The triad of renal cell carcinoma (hematuria, lumbar pain, and abdominal mass) is present in most patients with intermediate to advanced disease, and the presence of left spermatic varices suggests the possibility of left renal vein thrombosis; therefore, early diagnosis of renal cell carcinoma is important.
Paraneoplastic syndrome: The clinical manifestations are not directly caused by the primary tumor or the site of the metastasis, but by an abnormal immune response caused indirectly by the products secreted by the tumor or by other unexplained pathologies of the endocrine, neurological, digestive, hematopoietic, osteoarticular, renal, and cutaneous systems, with corresponding clinical manifestations. It is called paraneoplastic syndrome. The incidence of paraneoplastic syndrome in patients with renal cell carcinoma is about 30%, which manifests as hypertension, increased erythrocyte sedimentation rate, erythrocytosis, abnormal liver function, hypercalcemia, hyperglycemia, neuromuscular lesions, amyloidosis, overflow, and abnormal coagulation mechanisms. Patients who present with paraneoplastic syndrome have a worse prognosis.
Symptoms caused by metastatic foci: Some patients with renal cell carcinoma are affected by metastatic foci of
Clinical manifestations are the first symptoms to be seen, such as bone pain, fracture, cough, hemoptysis, etc.
Physical examination findings include enlarged cervical lymph nodes, secondary varicose veins and bilateral lower limb edema, the latter suggesting possible tumor invasion of the renal veins and inferior vena cava. Among patients with metastatic renal cell carcinoma, the common metastatic organs and the incidence of metastases were, in order of prevalence, pulmonary metastases (48.4%), bone metastases ( 23.2%), hepatic metastases (12.9%), adrenal metastases (5.2%), and skin metastases (1.9%), brain metastases (1.3%), other sites, etc. (1.3%), and other sites (7.1%). Patients with advanced disease may also present with symptoms of malignancy such as wasting, weakness, and poor nausea.
(ii) Laboratory tests.
The purpose of routine laboratory tests for renal cell carcinoma is to understand the general status of the patient and whether appropriate therapeutic measures are indicated, including urine routine, blood routine, erythrocyte sedimentation rate, blood glucose, blood calcium, renal function (blood urea nitrogen, blood creatinine and glomerular filtration rate), liver function, lactate dehydrogenase, alkaline phosphatase, etc. phosphatase, etc. If invasive testing or surgical treatment is required, the necessary coagulation tests should be performed. The results of the above tests may show hematuria, erythrocytosis, anemia, increased erythrocyte sedimentation rate, hyperglycemia, hypercalcemia, abnormal renal function and abnormal liver function in patients with renal cell carcinoma. For patients with renal tumors adjacent to or involving the renal pelvis, urine cytology examination is also required. Patients with renal tumors in isolated kidneys, bilateral renal tumors, abnormal renal function indicators and the presence of diseases that impair renal function (such as diabetes mellitus, chronic pyelonephritis, polycystic kidney, contralateral renal stone, etc.) need to undergo nuclear nephrography to understand renal function and to assess the grade of renal insufficiency. Currently, there are no accepted serum tumor markers for early adjuvant diagnosis of renal cell carcinoma.
(iii) Imaging.
With the popularity of imaging, now more than 50 of renal cell carcinomas are found unexpectedly during screening for nonspecific symptoms in the abdomen or disease in other organs. Imaging has an important role at different stages of the diagnosis and management of renal cell carcinoma: for the primary tumor lies in the detection, localization, characterization and staging of the lesion; it can assist in localization during surgery; and it is an important tool for follow-up during postoperative and nonoperative treatment.
of renal cell carcinomas are found unexpectedly during screening for nonspecific symptoms in the abdomen or disease in other organs. Imaging has an important role at different stages of the diagnosis and management of renal cell carcinoma: for the primary tumor lies in the detection, localization, characterization and staging of the lesion; it can assist in localization during surgery; and it is an important tool for follow-up during postoperative and nonoperative treatment.
⒈ chest X-ray
Patients with renal cell carcinoma should routinely undergo frontal and lateral chest X-rays, and patients with suspicious nodules on chest X-rays or clinical stage ≥ stage III should undergo chest CT.
PEC ultrasonography
Abdominal ultrasonography is the easiest and most common method to detect renal tumors. Renal ultrasonography is useful to identify benign and malignant renal tumors and is indicated for the differential diagnosis of patients with renal tumors in chronic renal failure or iodine allergy that precludes enhanced CT scanning, as well as patients with complex renal cysts.
- style=”margin-left: 107pt”>
- Diagnosis of primary foci of renal cell carcinoma.
-
Gray-scale versus Doppler ultrasound: ultrasound is economical, simple, radiation-free, with high prevalence, and is the preferred test for clinical suspicion of renal tumors. Most of the asymptomatic renal cell carcinoma is detected by ultrasound examination. Gray-scale ultrasound can show the size, location and relationship between tumor and surrounding tissues. Color Doppler flow imaging
(color Doppler flow imaging (CDFI) provides a picture of the status of the blood supply to the tumor and also provides a preliminary evaluation of venous tumor thrombosis. Gray-scale ultrasound and CDFI have a high sensitivity for the identification of cystic kidney tumors.
-
Ultrasound imaging: For solid renal tumors, enhanced imaging is one of the most important tools to identify benign and malignant lesions. Real-time gray-scale ultrasonography
(Contrast-enhanced ultrasound, CEUS) improves the sensitivity and accuracy of blood flow examination, provides more information on the early arterial perfusion and microcirculatory status of the mass, and is more sensitive and specific for detecting and showing features of renal cell carcinoma. CEUS is also highly sensitive and specific for the diagnosis of complex renal cysts.
-
Preoperative staging of renal cell carcinoma: ultrasound examination is limited in scope and influenced by imaging resolution, patient’s condition and operator’s experience. The accuracy of tumor staging is not as good as that of CT.
-
Intraoperative diagnosis of renal cell carcinoma: ultrasound is routinely used to guide tumor puncture biopsy because of its non-radiation, flexible and convenient features. The intraoperative examination can correctly visualize the kidney. The intraoperative examination can correctly visualize the renal tumor and provide a cleansing determination of the relationship between the tumor and the renal pelvis and the extent of the tumor thrombus in the renal vein, inferior vena cava, and right atrium.
- style=”margin-left: 48pt”>
- CT examination
Abdominal CT is the most common test for preoperative diagnosis and postoperative follow-up of renal cell carcinoma. CT scans can qualitatively diagnose most renal tumors and have high diagnostic sensitivity and specificity. On CT scan, renal clear cell carcinoma has a typical contrast “fast-in-fast-out” appearance: a heterogeneous iso-/low-density round-like mass on plain scan, and a medium- to highly-enhanced tumor in the dermal medullary phase and a low-density mass in the parenchymal phase. The tumor density in the parenchymal stage is lower than that in the renal parenchyma. However, the need for
- CT examination
Note that CT examination is useful for differentiating some rare types of renal cell carcinoma from benign tumors such as eosinophilic adenoma and lipid-depleted The difference between some rare types of renal cell carcinoma and benign tumors such as eosinophilic adenoma and lipid-depleted vascular smooth muscle lipoma remains difficult to distinguish.
In addition to the qualitative diagnosis, CT examination provides additional diagnostic information to the preoperative patient, including: the extent of tumor invasion, including whether the venous system is invaded (T stage), whether regional lymph nodes are metastatic (N stage), whether there is metastasis in the adjacent organs at the scan range (M stage), and whether there is metastasis in the adjacent organs at the scan range (M stage). The scope of tumor invasion, including whether the venous system is invaded (T-stage), whether regional lymph nodes are metastatic (N-stage), whether there is metastasis in organs adjacent to the scan range (M-stage), the presence of metastatic vessels (CTA) and a gross assessment of bilateral kidney morphology and function.
Bosniak classification of renal cystic masses: Renal cystic masses are a group of predominantly cystic diseases that can be congenital, infectious, secondary, or neoplastic (benign and malignant). The imaging presentation can range from simple cysts, slightly complex cystic lesions to complex cystic solid masses. bosniak classifies renal cystic masses into 4 categories based on CT presentation and provides clinical management according to the different levels (see Table 6 for details). The diagnosis of cystic masses is based on the CT presentation and provides clinical management according to the different classes (Table 6).
Table 6 Bosniak classification and management of renal cystic masses
Bosniak Classifieds |
CT Features |
Handle |
|
Class I |
①Simple cyst with thin and slender wall, no separation, calcification or solid components; ②Uniform watery density foci (CT value 0-20 HU); ③Lucid borders with smooth and sharp margins; ④No enhancement on enhancement scan |
Benign |
|
Class II |
①Benign cysts may be associated with slender separations; ②Tiny calcifications in the cyst wall or separations; ③<3 cm uniformly dense cysts; ④Sharp borders without enhancement |
Benign |
|
Class IIF |
①slightly increased fibrous compartments with slight uniform thickening and enhancement of the cyst wall or compartments; ②slightly thickened or nodular calcification within the cyst without enhancement; ③soft tissue component without enhancement; ④dense cysts ≥3 cm in diameter located entirely within the renal parenchyma. Usually well-defined |
Follow up to 5 years partially malignant |
Class III |
1) Difficult to characterize cyst with irregular or uniform increase in cyst wall or compartment
thickness; ②enhancement visible on enhanced scans |
Surgery or active follow-up.
More than 50 |
Class IV |
With typical signs of malignancy: there is an enhanced soft tissue component |
Surgery
Most are malignant |
 as malignant
as malignant
- style=”margin-left: 83pt”>
- MRI examination
Abdominal MRI is a more common test for preoperative diagnosis and postoperative follow-up of renal cell carcinoma, and can be used for patients who are allergic to CT contrast, pregnant women, or other patients who are not suitable for CT examination. MRI is more accurate than CT for the diagnosis of renal vein and inferior vena cava thrombosis, and it is clearer than CT for the structure within cystic lesions of the kidney.
Lucidly. The differential diagnosis of renal cell carcinoma and hemorrhagic renal cysts is also superior to CT, so MRI may be a better option than CT for these lesions.
- style=”margin-left: 83pt”>
- Positron emission tomography
At present, positron emission tomography-computed tomography (PET-CT) is the most widely used imaging agent in clinical practice. The most widely used imaging agent is fluorine-18-fluorodeoxyglucose.
(18F-fluorodeoxyglucose, 18F-FDG ), which is directly excreted by the kidney without metabolism after intravenous injection in about 50% of cases, can affect the display of renal lesions; on the other hand, the low expression of cell membrane GLUT-1 in grade I-II renal clear cell carcinoma and the excess of fluorodeoxyglucose-6-phosphate catabolic enzyme in renal cell carcinoma result in only about half of the primary foci of renal cell carcinoma showing increased metabolism of fluorodeoxyglucose, and the other half can be no different from normal renal parenchymal uptake. Therefore 18F-FDG PET-CT imaging has limited diagnostic value for primary foci of renal cell carcinoma and is not recommended for routine use. Other newer imaging agents that have been studied more frequently are fluorine-18 or carbon-11-labeled acetate, which have good imaging effects on well-differentiated, less malignant renal cell carcinoma and can compensate for the shortcomings of single 18F-FDG imaging, but they are still in the research stage and are not routinely used. However, several studies have also shown that PET-CT imaging is superior to conventional imaging methods for lymph node metastases and distant metastases in renal cell carcinoma, especially in determining bone metastases or skeletal muscle metastases in renal cell carcinoma, and is able to monitor the efficacy and predict the prognosis of patients early by changes in glucose metabolism.
- style=”margin-left: 83pt”>
- Nuclear bone imaging
Bone metastases from renal cell carcinoma preferably occur in the mid-shaft bone and the bone ends of long bones, either singly or in multiple cases, and mostly present as expansive, osteolytic bone destruction with early invasion of bone marrow tissue.
As the disease progresses, it destroys bone trabeculae, bone cortex, and forms soft tissue masses in the surrounding area. Nuclide bone imaging is the preferred screening method for bone metastases from renal cell carcinoma, but the sensitivity is only about 50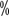 . Patients with bone related symptoms such as bone pain or elevated serum alkaline phosphatase or clinical stage ≥ III renal cell carcinoma should undergo bone scan to clarify whether there are bone metastases. The patient should have a bone scan to determine if there are bone metastases.
. Patients with bone related symptoms such as bone pain or elevated serum alkaline phosphatase or clinical stage ≥ III renal cell carcinoma should undergo bone scan to clarify whether there are bone metastases. The patient should have a bone scan to determine if there are bone metastases.
- style=”margin-left: 83pt”>
- Kidney dynamic imaging
Nuclear renal dynamic imaging can accurately evaluate preoperative bilateral and fractional kidney function in patients with renal cell carcinoma and help guide decisions about surgical options.
- style=”margin-left: 83pt”>
- Renal Tumor Puncture Biopsy
Percutaneous renal aspiration biopsy includes hollow-core needle biopsy and fine needle aspiration
(fine-needle aspiration, FNA), which can provide a pathologic histologic basis for renal tumors that cannot be diagnosed on imaging. A hollow-core needle biopsy has a higher accuracy than FNA for the diagnosis of malignant tumors. Hollow-core needle biopsy is preferred for renal tumors with solid components. The coaxial technique allows multiple biopsies to be taken through coaxial cannulae, avoiding the risk of potential tumor tract implantation and metastasis. At least two good quality tissue specimens should be obtained, avoiding necrotic areas. The diagnostic yield and accuracy of hollow-core needle biopsy for cystic renal tumors is low and is not recommended.
The risk of puncture and the potential risk of spread, although low, should not be ignored. Percutaneous
Renal puncture biopsy is not indicated in critically ill patients. Puncture biopsy is also not recommended for patients undergoing surgery because of the high diagnostic accuracy of enhanced abdominal imaging. For patients with renal cell carcinoma who are not suitable for surgical treatment (old and frail, or have contraindications to surgery), or patients with advanced renal cell carcinoma who cannot be treated surgically, a puncture biopsy of renal tumor before systemic systemic therapy to clarify the pathological diagnosis (including the type of pathology) can help to select the therapeutic drugs. Patients with renal cell carcinoma who choose ablation therapy should have renal tumor aspiration biopsy to obtain pathological diagnosis. Therefore, in practice, it is still necessary to consider the risk of puncture, the skill level of the operator, and whether it may affect the current treatment plan to make a comprehensive decision.
V. Prognostic evaluation of advanced/metastatic renal cell carcinoma
Prognostic risk models for advanced renal cell carcinoma are useful for patient risk stratification and treatment selection, and are commonly used today, including the Memorial Sloan Kettering Cancer Center (MSKCC) criteria and the International Metastatic Cancer Center (IMCC) criteria. The MSKCC score is based on the cytokine era, including physical status, lactate dehydrogenase, hemoglobin, calcium, and self-diagnosis to systemic risk (Table 7). The MSKCC score was established in the cytokine era and included 5 risk factors: physical status, lactate dehydrogenase, hemoglobin, blood calcium, and time from diagnosis to systemic therapy, classified as low, intermediate, and high risk, corresponding to intermediate
The median overall survival time was 30 months, 14 months, and 5 months. The IMDC score applied in the era of targeted therapy, built on the MSKCC criteria, included 4 of the MSKCC prognostic factors (except lactate dehydrogenase), and incorporated platelet and neutrophil counts, with median overall survival times for low-risk, intermediate-risk, and high-risk patients, respectively
35.3 months, 16.6 months, and 5.4 months.

Table 7 Prognostic risk assessment criteria for advanced renal cell carcinoma
Risk factors MSKCC criteria IMDC criteria


- style=”margin-left: 95pt”>
- Diagnosis-to-treatment interval <1 year Diagnosis-to-treatment interval <1 year
- style=”margin-left: 95pt”>
- Kanofsky Count Score <80

Kanofske count <80
- style=”margin-left: 95pt”>
- Serum calcium > upper limit of normal index Serum calcium > upper limit of normal index
- style=”margin-left: 95pt”>
- Hemoglobin
- style=”margin-left: 95pt”>
- Lactate dehydrogenase > upper limit of normal index 1.5 times Neutrophil > upper limit of normal index
- style=”margin-left: 287pt”>
 platelet levels > upper limit of normal index
platelet levels > upper limit of normal index
Risk grouping


Low risk group 0 risk factors 0 risk factors
Medium risk group 1 to 2 risk factors 1 to 2 risk factors
 < span style="font-size:12pt">High risk group 3 to 5 risk factors 3 to 6 risk factors
< span style="font-size:12pt">High risk group 3 to 5 risk factors 3 to 6 risk factors
VI.
Patients with renal cell carcinoma have their clinical staging determined by imaging, and their ability to tolerate treatment is assessed using ancillary tests. For patients undergoing surgery, pathologic staging is determined based on pathologic findings, and postoperative treatment and follow-up options are selected based on pathologic staging.
(i) Surgical treatment.
Surgery remains the treatment of choice for patients with limited and locally progressive renal cell carcinoma that may result in a cure. For patients with elective advanced renal cell carcinoma, if the patient is able to tolerate surgical treatment in a systemic
Decompensated nephrectomy and isolated metastasis resection in addition to systemic therapy may also improve patient survival.
- style=”margin-left: 83pt”>
- RN
In 1963 Robson et al. established the basic principles of RN and established it as the gold standard for the surgical management of limited renal cell carcinoma. The classic scope of RN resection included the affected kidney, perirenal fascia, perirenal fat, ipsilateral adrenal glands, lymph nodes from the foot of the diaphragm to the bifurcation of the abdominal aorta, and the ureter above the bifurcation of the iliac vessels. Current concepts have changed, and intraoperative adrenalectomy and regional lymph node dissection are not recommended routinely.
- style=”margin-left: 83pt”>
- Preservation of renal unit surgery
Patients left with only one kidney after RN may have decreased kidney function and increased risk of chronic renal insufficiency and dialysis. Chronic renal insufficiency increases the patient’s risk of cardiovascular events and increases overall mortality. For patients with limited renal cell carcinoma, nephron sparing surgery (NSS) is recommended for patients with clinical stage T1a renal cell carcinoma, if technically feasible. The thickness of the normal renal parenchyma surrounding the tumor to be removed is not a critical issue, as long as the final surgical specimen has negative margins. Although there is an increased risk of local tumor recurrence after partial nephrectomy, the patient-specific mortality rate is similar to that of RN. The location of the tumor (exophytic or endophytic) is more important than the size of the tumor for the feasibility of partial nephrectomy. A tumor that is too large or too deeply located increases the time of thermal ischemia during renal surgery and the risk of complications from postoperative bleeding and urinary leakage increases. Therefore, the indications for NSS are also to some extent
depends to some extent on the surgeon’s experience and surgical technique.3. Surgery-related issues
-
Open surgery / laparoscopic surgery / robot-assisted technology: Compared to traditional open surgery, the advantages of laparoscopic surgery are Small surgical incision, less injury, less bleeding, faster postoperative recovery, fewer comorbidities, shorter hospital stay, and recent tumor control rate is not significantly different from that of open surgery. The disadvantages are expensive instruments, complex technique, long learning curve for proficiency, and long operative time in the initial stage. As the technique becomes more proficient, the operative time will be significantly reduced and the degree of complete resection will be exactly the same as that of open surgery. The introduction of the da Vinci robot has made several key steps of laparoscopic partial nephrectomy easier to master and the learning curve faster. Currently, open surgery, laparoscopic surgery, or robotic-assisted techniques can all be used for the surgical treatment of patients with renal cell carcinoma when technology allows, and the choice depends largely on the size and location of the renal tumor and the surgeon’s level of experience.
-
Ipsilateral adrenalectomy: The classic scope of RN includes the ipsilateral adrenal gland. However, given the low risk of ipsilateral adrenal involvement in smaller renal cell carcinomas, intraoperative preservation of the ipsilateral adrenal gland should be considered in the absence of an abnormal adrenal gland on CT scan. If ipsilateral adrenal abnormalities are found during surgery, they should be removed.
-
Regional lymph node dissection: The need for retroperitoneal regional lymph node dissection in the setting of RN is also controversial. There is controversy. There is no evidence that lymph node dissection is beneficial to patients. The European organization for research and treatment of cancer (EORTC) is a leading organization in the field of lymph node dissection.
The results of a 20-year randomized controlled phase III clinical study by the European Organization for Research and Treatment of Cancer (EORTC) showed that in resectable limited renal cell carcinoma (RCC) (
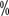
 , mostly due to multifocal or positive cut margins of the primary renal cell carcinoma. There is controversy as to whether a positive surgical margin for partial nephrectomy increases the patient’s risk of tumor recurrence and its prognostic impact. Studies have shown that even with positive margins for partial nephrectomy, there is no increase in tumor recurrence at midterm follow-up. Even some studies have shown no evidence of residual tumor in the vast majority of patients undergoing remedial nephrectomy immediately after surgery or later. Literature Reports 3
, mostly due to multifocal or positive cut margins of the primary renal cell carcinoma. There is controversy as to whether a positive surgical margin for partial nephrectomy increases the patient’s risk of tumor recurrence and its prognostic impact. Studies have shown that even with positive margins for partial nephrectomy, there is no increase in tumor recurrence at midterm follow-up. Even some studies have shown no evidence of residual tumor in the vast majority of patients undergoing remedial nephrectomy immediately after surgery or later. Literature Reports 3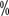
 of NSS will have positive postoperative pathological margins, but only those patients with higher pathological grading (grade III-IV) will have an increased risk of postoperative recurrence.
of NSS will have positive postoperative pathological margins, but only those patients with higher pathological grading (grade III-IV) will have an increased risk of postoperative recurrence.. Because surgical treatment of venous aneurysms is associated with greater risk and complications, preoperative evaluation requires thorough preparation, a detailed treatment plan, and an experienced team to perform the procedure.</p>
</li>
</ul>
<p style=)




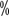 Patients showed a downstaging of their staging, with The pathological stage was T2 in 2 patients. Multifactorial regression analysis showed that pT4 and lymph node metastasis were independent predictors of survival prognosis. The 3-year overall survival rate for lymph node negative patients was 66
Patients showed a downstaging of their staging, with The pathological stage was T2 in 2 patients. Multifactorial regression analysis showed that pT4 and lymph node metastasis were independent predictors of survival prognosis. The 3-year overall survival rate for lymph node negative patients was 66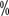 , while the 3-year overall survival rate for patients with lymph node metastasis was 12
, while the 3-year overall survival rate for patients with lymph node metastasis was 12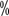 for lymph node negative patients and 12
for lymph node negative patients and 12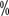 positive margins). Survival time was significantly shorter in patients with positive margins. The median survival time for the entire group of patients was
positive margins). Survival time was significantly shorter in patients with positive margins. The median survival time for the entire group of patients was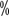 . However, in patients with lymph node metastases, no significant benefit was seen. Multidisciplinary collaboration is important in the management of patients with clinical stage T4 renal cell carcinoma, as it involves the resection and reconstruction of adjacent organs. In summary, aggressive surgery in patients with clinical T4N0M0 renal cell carcinoma may provide a significant benefit if conditions allow.
. However, in patients with lymph node metastases, no significant benefit was seen. Multidisciplinary collaboration is important in the management of patients with clinical stage T4 renal cell carcinoma, as it involves the resection and reconstruction of adjacent organs. In summary, aggressive surgery in patients with clinical T4N0M0 renal cell carcinoma may provide a significant benefit if conditions allow.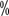 and 96
and 96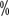 and 75
and 75 (
(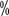 and 65.9
and 65.9 to 100
to 100 ,
,  to 100
to 100 ,
, 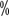
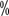 , 5-year tumor-specific mortality 0.2
, 5-year tumor-specific mortality 0.2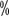
 , 5-year progression-free survival rate 97
, 5-year progression-free survival rate 97 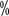
 . AS is a viable option for patients with older or frail SRMs. American Urologic Surgery
. AS is a viable option for patients with older or frail SRMs. American Urologic Surgery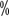 to 40
to 40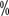 , with 8
, with 8 ~16
~16  of patients with grade III or higher hypertension. The incidence reported in China is similar to that abroad, with an incidence of all grades of hypertension ranging from 15
of patients with grade III or higher hypertension. The incidence reported in China is similar to that abroad, with an incidence of all grades of hypertension ranging from 15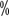 to 37 < img src="https://www.kiraspecialist.com/wp-content/uploads/2022/06/062222_1307_202265.png" alt=""/>. Baseline blood pressure should be assessed before starting targeted therapy, and for patients with pre-existing hypertension the target blood pressure should be controlled below 140/90 mmHg during treatment. When hypertension reaches grade II or higher or grade I with symptoms, it must be controlled with medication. The best choice of antihypertensive medication is blood
to 37 < img src="https://www.kiraspecialist.com/wp-content/uploads/2022/06/062222_1307_202265.png" alt=""/>. Baseline blood pressure should be assessed before starting targeted therapy, and for patients with pre-existing hypertension the target blood pressure should be controlled below 140/90 mmHg during treatment. When hypertension reaches grade II or higher or grade I with symptoms, it must be controlled with medication. The best choice of antihypertensive medication is blood


 < span style="font-size:12pt">V Level Death.
< span style="font-size:12pt">V Level Death.
 < span style="font-size:10pt">V – – Death
< span style="font-size:10pt">V – – Death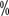 , with an incidence of 16.1
, with an incidence of 16.1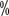 for grade ≥III HFS. Cutaneous reactions in the hands and feet are more common in Chinese patients, with the literature reporting an incidence of 55
for grade ≥III HFS. Cutaneous reactions in the hands and feet are more common in Chinese patients, with the literature reporting an incidence of 55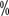 for all levels of HFS > to 68
for all levels of HFS > to 68 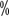 . Clinical manifestations of skin toxicity are dry skin, rash, pruritus, blistering, molting, localized thickening of skin keratin, or seborrheic dermatitis with skin sagging. It usually appeared 3 to 8 weeks after the start of treatment. In targeted therapy, the incidence of all graded rashes was 13
. Clinical manifestations of skin toxicity are dry skin, rash, pruritus, blistering, molting, localized thickening of skin keratin, or seborrheic dermatitis with skin sagging. It usually appeared 3 to 8 weeks after the start of treatment. In targeted therapy, the incidence of all graded rashes was 13 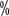 to 37
to 37  ~4.0
~4.0  Examine the palms and soles of the feet prior to treatment to exclude pre-existing areas of skin keratinization. Intervene immediately when symptoms appear with an ointment or lotion containing a 10
Examine the palms and soles of the feet prior to treatment to exclude pre-existing areas of skin keratinization. Intervene immediately when symptoms appear with an ointment or lotion containing a 10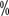 urea component; if hyperkeratosis occurs, then use an oil or lotion containing
urea component; if hyperkeratosis occurs, then use an oil or lotion containing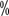 to 40
to 40 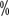 Urea for exfoliation treatment. For grade II or higher, use an ointment containing 0.05
Urea for exfoliation treatment. For grade II or higher, use an ointment containing 0.05 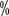 clobetasol; for pain, use a topical analgesic such as 2
clobetasol; for pain, use a topical analgesic such as 2 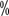 lidocaine may be used if pain is present. If severe symptoms occur, a dermatologic consultation is recommended. When grade II or higher HFS occurs, consider interrupting dosing until symptom severity resolves to less than grade I. Reduce or restart treatment at the same dose.
lidocaine may be used if pain is present. If severe symptoms occur, a dermatologic consultation is recommended. When grade II or higher HFS occurs, consider interrupting dosing until symptom severity resolves to less than grade I. Reduce or restart treatment at the same dose.


 body surface area
body surface area body surface area
body surface area < span style="font-size:12pt">Class V – Death
< span style="font-size:12pt">Class V – Death
 ~
~ There were varying degrees of hypothyroidism (Table 13), and the incidence increased progressively with the duration of treatment. The results of the domestic study showed a slightly higher incidence of hypothyroidism than in the Western population, ranging from 14.0
There were varying degrees of hypothyroidism (Table 13), and the incidence increased progressively with the duration of treatment. The results of the domestic study showed a slightly higher incidence of hypothyroidism than in the Western population, ranging from 14.0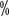 to 24.9
to 24.9 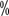 . Transient hyperthyroidism may occur in some patients, usually without intervention, and most will progress to hypothyroidism on subsequent treatment. The classification of hypothyroidism for targeted therapy in renal cell carcinoma is shown in Table 5. thyroid function tests are performed at the beginning of treatment and thyroxine and thyroid stimulating hormone (TSH) are monitored closely during targeted therapy. Patients with mildly elevated TSH without symptoms need only continued monitoring, while patients with TSH>10 mU/L or clinical signs of hypothyroidism require thyroid hormone replacement therapy. In most cases, thyroid hormone replacement therapy is effective in controlling symptoms and does not require suspension of targeted drug therapy or dose adjustment.
. Transient hyperthyroidism may occur in some patients, usually without intervention, and most will progress to hypothyroidism on subsequent treatment. The classification of hypothyroidism for targeted therapy in renal cell carcinoma is shown in Table 5. thyroid function tests are performed at the beginning of treatment and thyroxine and thyroid stimulating hormone (TSH) are monitored closely during targeted therapy. Patients with mildly elevated TSH without symptoms need only continued monitoring, while patients with TSH>10 mU/L or clinical signs of hypothyroidism require thyroid hormone replacement therapy. In most cases, thyroid hormone replacement therapy is effective in controlling symptoms and does not require suspension of targeted drug therapy or dose adjustment.


 < span style="font-size:12pt">V Death
< span style="font-size:12pt">V Death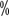 . The use of
. The use of ~10
~10  , as evidenced by decreased left ventricular ejection fraction (1eft ventricular ejection fraction (LVEF)) and myocardial ischemia. In patients without cardiac risk factors, baseline LVEF testing should be considered. Patients with cardiac risk factors or recent cardiovascular adverse events should be closely monitored for vital signs and LVEF. target therapy should be suspended if congestive heart failure occurs; if symptomatic congestive heart failure does not occur, but LVEF<50
, as evidenced by decreased left ventricular ejection fraction (1eft ventricular ejection fraction (LVEF)) and myocardial ischemia. In patients without cardiac risk factors, baseline LVEF testing should be considered. Patients with cardiac risk factors or recent cardiovascular adverse events should be closely monitored for vital signs and LVEF. target therapy should be suspended if congestive heart failure occurs; if symptomatic congestive heart failure does not occur, but LVEF<50 , or a decrease in LVEF of 20
, or a decrease in LVEF of 20 , the targeted drug dose should be reduced or treatment suspended. Patients with a previous history of long Q-T intervals, antiarrhythmic drugs, bradycardia, and electrolyte abnormalities should have regular electrocardiograms and blood potassium and magnesium tests.
, the targeted drug dose should be reduced or treatment suspended. Patients with a previous history of long Q-T intervals, antiarrhythmic drugs, bradycardia, and electrolyte abnormalities should have regular electrocardiograms and blood potassium and magnesium tests.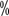 . A growing number of studies have shown that in most stage T1, some stage T2, and even some stage T3a renal cell carcinomas, partial nephrectomy is associated with
. A growing number of studies have shown that in most stage T1, some stage T2, and even some stage T3a renal cell carcinomas, partial nephrectomy is associated with



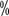 < span style="font-family:Times New Roman">
< span style="font-family:Times New Roman">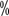 ) and higher rates of complete remission (16.1
) and higher rates of complete remission (16.1 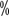
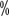 ). The ≥3 grade treatment-related adverse reactions were 71.6
). The ≥3 grade treatment-related adverse reactions were 71.6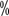 and 58.8
and 58.8

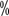
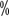 ).
).
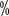 , P<0.001) and median overall survival There were significant benefits in terms of time (not reached vs. 26 months, P<0.001). Based on the results of this study, in April 2018 the US Food and Drug Administration approved nabritumomab in combination with epirimizumab as standard first-line therapy for high-risk advanced renal cell carcinoma in IMDC.
, P<0.001) and median overall survival There were significant benefits in terms of time (not reached vs. 26 months, P<0.001). Based on the results of this study, in April 2018 the US Food and Drug Administration approved nabritumomab in combination with epirimizumab as standard first-line therapy for high-risk advanced renal cell carcinoma in IMDC.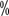 of whom were at low to moderate risk for MSKCC, with median progression-free survival times of 11 months and 5 months (HR 0.42.
of whom were at low to moderate risk for MSKCC, with median progression-free survival times of 11 months and 5 months (HR 0.42.
 and
and (P<0.001), with median survival times of 26.4 months and 21.8 months, respectively
(P<0.001), with median survival times of 26.4 months and 21.8 months, respectively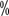
 , 61
, 61 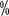 and 41
and 41 
 and 9
and 9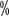 (P=0.0001), respectively.
(P=0.0001), respectively. crossed over to the everolimus group, so there was no significant difference in median survival time between the two groups, which was 14.8
crossed over to the everolimus group, so there was no significant difference in median survival time between the two groups, which was 14.8 and 39.0
and 39.0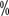 , the sorafenib group had better quality-of-life scores and was better tolerated. Due to the lack of validated large studies of sorafenib in first-line therapy and the increasing availability of alternatives, the NCCN guidelines do not currently recommend sorafenib for first-line treatment of renal clear cell carcinoma, but mainly for post-treatment. A national multicenter study of 845 patients with advanced renal cell carcinoma in the first line
, the sorafenib group had better quality-of-life scores and was better tolerated. Due to the lack of validated large studies of sorafenib in first-line therapy and the increasing availability of alternatives, the NCCN guidelines do not currently recommend sorafenib for first-line treatment of renal clear cell carcinoma, but mainly for post-treatment. A national multicenter study of 845 patients with advanced renal cell carcinoma in the first line , with a median progression-free survival time of 8.7 months. Similarly, in 2006 JAMA reported a retrospective study of 106 patients with an effective rate of 34
, with a median progression-free survival time of 8.7 months. Similarly, in 2006 JAMA reported a retrospective study of 106 patients with an effective rate of 34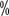 with a median progression-free survival time of 8.3 months.
with a median progression-free survival time of 8.3 months.
 and 3
and 3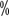 , respectively.
, respectively. and 5
and 5 , with median progression-free survival times of 4.6 months and 4.4 months, respectively. 3/4 degree adverse events occurred at 19
, with median progression-free survival times of 4.6 months and 4.4 months, respectively. 3/4 degree adverse events occurred at 19 and 37
and 37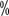 , respectively.
, respectively. , median progression-free survival time
, median progression-free survival time .
.

 , with a median progression-free survival time
, with a median progression-free survival time , with a median progression-free survival time of 10.6 months. 108 patients with primary non-clear cell carcinoma in the ASPEN study were randomized to
, with a median progression-free survival time of 10.6 months. 108 patients with primary non-clear cell carcinoma in the ASPEN study were randomized to , with a median progression-free survival time of 8.6 months and a median survival time of 19.6 months.
, with a median progression-free survival time of 8.6 months and a median survival time of 19.6 months. , disseminated papillary carcinoma efficiency 29
, disseminated papillary carcinoma efficiency 29 , median progression-free survival time minutes
, median progression-free survival time minutes .
. , disseminated papillary carcinoma efficiency 29
, disseminated papillary carcinoma efficiency 29 , and median progression-free Survival times were
, and median progression-free Survival times were .
. , effective rate 27
, effective rate 27 , median progression-free survival time and overall survival time were
, median progression-free survival time and overall survival time were span style=”font-family:MS UI Gothic”>∼1 year
span style=”font-family:MS UI Gothic”>∼1 year , with a 1-year pain control rate of 82
, with a 1-year pain control rate of 82  , with only 0
, with only 0 .
.