Hemophilia A Guidelines
(2022 Edition)
Hemophilia A (HA) is an inherited bleeding disorder with X-chromosome linked recessive inheritance. The main clinical manifestation is a qualitative or quantitative abnormality of coagulation factor VIII (FVIII). The clinical manifestations are characterized by bleeding that is difficult to stop after spontaneous or minor trauma to joints, muscles, viscera, and deep tissues, often starting in childhood. The prevalence of HA is about 1 in 5,000 men, and hemophilia is extremely rare in women. The prevalence of hemophilia in China is 2.73/100,000 population, of which HA accounts for 80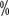 ~The prevalence of haemophilia is 2.73 per 100,000 population, with HA accounting for 80
~The prevalence of haemophilia is 2.73 per 100,000 population, with HA accounting for 80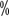 .
.
HA is a disease caused by deficiency or abnormal quality of a single clotting factor. Early recognition and diagnosis of hemophilia allows patients to live a normal life by avoiding bleeding and the joint damage and disability caused by bleeding through proper and correct preventive treatment or timely replacement therapy after bleeding.
Due to the important role of FⅧ in the endogenous coagulation pathway, patients with HA The clinical presentation is characterized by bleeding that can occur anywhere in the body. The most common sites of bleeding are joints, muscles, and deep tissues, but also the gastrointestinal , urinary , central nervous system bleeding, and after tooth extraction bleeding, etc. If left untreated it can lead to joint deformities and pseudotumors, etc., and can even be life-threatening in severe cases. Persistent bleeding after trauma or surgery is also a feature of this disease.
The degree of bleeding correlates with FVIII activity. Activity correlates with lighter patients, who generally bleed rarely and only after injury or surgery; heavy patients, who bleed from an early age and can bleed from any part of the body; and intermediate patients, who bleed to a degree of severity between light and heavy.
A bleeding disorder, including hemophilia, should be considered when a male patient, especially a child, presents with spontaneous bleeding, or bleeding that does not stop after trauma or surgery. Follow up with the patient’s family history and further refine laboratory tests to confirm the diagnosis.
(i) Screening test.
Including platelet count, peripheral blood smear (platelet morphology), prothrombin time (PT), activated partial thromboplastin time (APTT) Patients with HA show only prolonged APTT, but some patients with mild HA have only mildly prolonged or high limit of normal APTT. The platelet count and morphology, as well as other coagulation parameters, should be normal.
(ii) Confirmatory tests.
Clotting factor testing: A prolonged APTT suggests an abnormal endogenous coagulation process, and coagulation markers associated with this should be examined, including FVIII, FIX, FXII activity, and vascular hemophilia factor antigen (VWF:Ag).
VIII:C) is reduced or absent, VWF:Ag is normal, and FVIII:C/VWF:Ag is markedly reduced, suggesting HA.
(iii) Inhibitor assay.
1.
Patients with HA should be tested for FⅧ inhibitors when they have a lower treatment effect than before and before they undergo surgery. In pediatric patients, testing is recommended every 5 exposure days for the first 20 exposure days after initial treatment with F VIII products and every 21 to 50 exposure days after initial treatment with F VIII products.
every 10 exposure days for the first 20 exposure days and at least 2 times per year thereafter.
up to 150 exposure days. 2. Inhibitor screening.
An APTT correction test was performed, in which normal plasma and patient plasma were mixed 1:1, incubated immediately and at 37°C for 2 hours, and then the APTT was measured and compared with the APTT of normal subjects and the patient itself, and corrected immediately. If the incubation is not corrected at 2 hours, the presence of coagulation factor inhibitors is indicated (see Table 1 for interpretation of the APTT correction test).
3. Inhibitor titer.
Inhibitor titers must be measured to confirm the diagnosis of inhibitor. A patient is considered positive if he/she has an inhibitor titer ≥0.6 BU/ml on 2 consecutive occasions within 1 to 4 weeks by the Bethesda or Nijmegen method. If the inhibitor titer >5 BU/ml, the inhibitor is a high titer inhibitor; if the inhibitor titer is ≤5 BU/ml, the inhibitor is a low titer inhibitor.
(iv) Genetic testing.
Genetic testing of patients is recommended to identify the causative gene and to provide a basis for carrier testing and prenatal diagnosis in the same family. In addition, mutations can be used to determine a patient’s risk of developing a suppressor.
According to the activity level of FⅧ, HA can be classified into mild, moderate, and heavy types (see ). medium and heavy types (see Table 2). Factor activity <1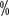 is considered heavy; activity 1
is considered heavy; activity 1 to 5
to 5  for medium; activity >5
for medium; activity >5  to 40
to 40 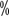 for light.
for light.
(a) Diagnosis.
Based on the patient’s clinical presentation with severe recurrent bleeding since childhood, especially joint bleeding, combined with FVIII factor The diagnosis is confirmed by laboratory tests for decreased activity and antigen with mutations in the FⅧ gene.
(ii) Differential diagnosis.
- Vascular hemophilia (VWD): VWD is an autosomal inherited disease due to a qualitative or quantitative defect in vWF. It is mostly dominant. Patients have a tendency to bleed, but mainly from the skin and mucous membranes. Because vWF has the ability to increase the stability of FVIII:C, prevent its degradation, and promote its production and release, patients with VWD may develop FⅧdecreased activity. Therefore, patients with decreased FVIIIactivity need to be excluded from VWD, especially in women. vWF antigens and activity (ristocetin cofactor activity, vWF cofactor activity) are required for the diagnosis and staging of VWD. VWD can be diagnosed and typed with vWF antigen and activity (ristomycin cofactor activity, vWF R:Co), collagen binding assay, FVIIIbinding assay, platelet adhesion and aggregation assay, vWF protein electrophoresis, etc. vWF protein electrophoresis, etc. Genetic diagnosis is also a diagnostic tool.
-
Acquired HA: is the presence of circulating anti-FVIIIautoantibodies in the circulation leading to FⅧa form of autoimmune disease with decreased activity that needs to be differentiated from HA, especially in patients with HA combined with suppressors. Acquired HA is characterized by no previous history of bleeding or positive family history and occurs most often in patients with malignancy, autoimmune disease, and perinatal women, although approximately half of patients have no apparent trigger and are effectively treated with immunosuppression.
-
Other inherited coagulation factor deficiencies: Patients found to have a prolonged APTT alone should also be tested for the activity of coagulation factors such as F Ⅸ, F Ⅺ, F Ⅻ, F Ⅴ, and F Ⅹ that can cause a prolonged APTT when performing confirmatory tests to rule out the corresponding coagulation factor deficiency disorders.
(a) Principles of treatment.
Patients with HA require replacement therapy with FⅧ and regular replacement therapy (prophylaxis) in the absence of bleeding, with the aim of stopping bleeding and thus maximizing preservation of joint function. The aim is to stop bleeding and thus maximize joint function. If bleeding occurs, adequate replacement therapy should be given promptly and on demand, and when surgery or other traumatic operations are performed, adequate replacement therapy should be administered. Patients with HA should avoid intramuscular injections and trauma.
(ii) Replacement therapy.
- style=”margin-left: 48pt”>
- Alternative therapy drug selection.
Recombinant F VIII or virally inactivated blood-derived F VIII is preferred for HA replacement therapy, and cold precipitation or fresh frozen plasma can be used when these drugs are not available. Each infusion of 1 IU/kg body weight of FⅧ can lead to an increase in FⅧ activity in the body.
(FⅧ:C) by 2% and the required FVIII in the body is 8-12 hours, and it is necessary to keep the FVIII:C in the body at a certain level. C should be infused every 8 to 12 hours to keep the body’s FVIII:C level at a certain level.
FVIII first requirement = (FVIII required to be achieved) Roman”>Ⅷ concentration– patient’s basal FⅧ concentration The patient’s basal F× weight (kg)× 0.5; after the first dose, half of the first dose may be infused every 8 to 12 hours as appropriate After the first dose, half of the first dose may be infused every 8 to 12 hours as appropriate until complete hemostasis.
Recovery rates and half-lives vary widely among individuals, and it is recommended that units that have the capacity to do so review the dose.
- Alternative therapy drug selection.
Measure pharmacokinetic parameters, such as recovery and half-life, in patients and guide therapy based on the results.
- style=”margin-left: 59pt”>
- Implementation of alternative therapies.
Alternative treatment is divided into on-demand treatment and regular alternative treatment (preventive treatment).
-
On-demand treatment: refers to the treatment of patients with acute bleeding or when the most effective current The most effective hemostatic measure remains FVIIIreplacement therapy, with the principle of early, adequate, and full course of treatment. Replacement therapy FVIIIdose and duration should take into account the site and severity of bleeding (see Table 3).
-
Perioperative replacement therapy: refers to replacement therapy before, during, and after surgery. The purpose is to ensure the smooth implementation of surgery and the smooth recovery of HA patients after surgery. See Exhibit 4 for specific alternative treatment options.
-
Prophylactic therapy: is a regular replacement therapy given periodically to prevent bleeding. Since on-demand treatment is only post-bleeding and cannot prevent recurrent bleeding leading to joint disability in patients with severe HA, prophylactic treatment is particularly critical as the goal is to maintain normal joint and muscle function. Prophylactic therapy is the treatment of choice for pediatric HA patients. A goal of less than 3 joint bleeds per year should be set for pediatric patients to minimize the occurrence of joint damage and irreversible joint disability due to joint bleeding.
There is no consensus on adherence to prophylaxis in adult patients, but experience both nationally and internationally has demonstrated that short-term tertiary prophylaxis can reduce the number of bleeds and improve quality of life. In addition, for patients with recent bleeding exacerbations, particularly in target joints where the frequency of bleeding has increased, short-term prophylaxis for 4 to 8 weeks is recommended to interrupt the vicious cycle of bleeding-joint injury. This treatment can be combined with intensive physical therapy or radiation therapy.
Radiation synovectomy.
There are usually three types of prophylactic treatment: 1) primary prophylaxis: after diagnosis, before the second joint bleed, and in children younger than 3 years of age with no clear evidence
Regular continuous replacement therapy is initiated when the presence of an arthropathy is confirmed (on examination or imaging); (ii) Secondary prevention therapy: Regular continuous replacement therapy is initiated after two or more bleeds in the joint, but no arthropathy is found on examination and/or imaging; (iii) Tertiary prevention therapy: Regular continuous replacement therapy is initiated after two or more bleeds in the joint, but no arthropathy is found on examination and/or imaging; and (iv) Tertiary prevention therapy. (3) Tertiary prophylaxis: regular continuous replacement therapy is started only after the presence of arthropathy is confirmed by physical examination and imaging.
To prevent joint disability, it is recommended that prophylaxis be initiated in children with severe disease after the first joint bleed, severe muscle bleed, intracranial bleed, or other life-threatening bleed. Children with a history of joint bleeding and arthropathy should start prophylaxis early, depending on their condition, and achieve a goal of <3 joint bleeds per year or <3 bleeds per year if possible.

③Small dose regimen: 10-15 IU/kg per dose, 2 to 3 times per week; followed by
30 IU/kg twice a week; followed by 25 IU/kg every other day. The pharmacokinetic-guided prophylaxis profile: individual pharmacokinetic parameters are used to develop a prophylactic regimen based on the patient’s actual needs, which allows for optimal dose and frequency of therapy and optimal resource allocation compared to a high-dose regimen, while maintaining efficacy.
While the optimal prophylaxis regimen remains to be determined, a low-dose regimen may significantly reduce bleeding in children with hemophilia compared with on-demand therapy, but does not reduce the incidence of arthropathy. It is recommended that medium-dose prophylaxis regimens be implemented in children with hemophilia who are financially able to do so, or that individualized regimens be optimized based on age, venous access, bleeding phenotype, pharmacokinetic characteristics, and availability of coagulation factor preparations.
(iii) Non-factor therapy.
-
Emicizumab: a bispecific monoclonal antibody that mimics F. family:Times New Roman”>VIIIa by mimicking the cofactor function of FVIIIa, which can simultaneously bridge the FVIIIa span style=”font-family:Arial”>a and FX, making FXin the absence of FVIII. span>is able to continue activation in the absence of F, restoring the physiological coagulation pathway. It is approved for routine prophylactic treatment of patients with HA in combination with FVIIIinhibitors in China and in the US and EU without Finhibitors. span>VIIIinhibitors in the United States and the European Union, and for routine prophylactic treatment of HA patients without Finhibitors in the United States and the European Union. The recommended dosing regimen is a loading dose of 3 mg/kg administered subcutaneously once weekly for the first 4 weeks to rapidly achieve target blood concentrations, and a dose of 3 mg/kg for the first 4 weeks.
A maintenance dose of 1.5 mg/kg once weekly was given starting in week 5.
-
Deamino-8-D-arginine pressor (DDAVP): light HA DDAVP is optional in patients with bleeding, and may be effective in a few intermediate HA types but not in patients with heavy HA. The recommended dose is 0.3-0.4μg/kg, diluted in 50 ml of physiological saline and administered slowly intravenously (at least 30 minutes) every 12 hours. The dose is administered once every 12 hours for 1 to 3 days. Increase in coagulation factor concentration >30
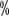 or higher after use
or higher after use>3-fold is considered effective. The drug has poor efficacy after multiple doses and should be supplemented with FVIII preparations in case of poor results. Adverse reactions include temporary flushing and sodium retention. Due to adverse reactions such as sodium and water retention, the
It is contraindicated in children under 2 years of age. Water restriction and pre-testing are required for use in young children. Children with a valid pretest may also use DDAVP nasal spray, which is designed for use in patients with hemophilia, to control minor bleeding.
- Antifibrinolytic drugs: Commonly used drugs include tranexamic acid, 6-aminohexanoic acid, and aminolevulinic acid. These drugs are effective for bleeding in the mouth, tongue, tonsils, throat and bleeding caused by tooth extraction, but they are less effective for bleeding in joint cavities, deep muscles and internal organs. Dosage: 6-Aminohexanoic acid 50-100mg/kg every 8-12 hours; tranexamic acid 10mg/kg intravenously or 25mg/kg orally; aminolevulinic acid 2-6mg/kg every 8 hours. It can also be used as a mouth rinse, especially during tooth extraction and oral bleeding, 5
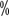 of tranexamic acid solution 10ml Rinse for 2 minutes, 4 times a day for 7 days.
of tranexamic acid solution 10ml Rinse for 2 minutes, 4 times a day for 7 days. -
Analgesic treatment: acetaminophen or opioid depending on the degree of pain, or COX-2 The pain is treated with acetaminophen or opioids, or COX-2 analgesics. In principle, aspirin or other non-steroidal analgesics are contraindicated, as well as all drugs that may affect platelet function.
(iv) Physical therapy.
Patients are encouraged to perform appropriate, safe aerobic exercise (swimming, power cycling, jogging, brisk walking, etc.) with appropriate load resistance strength training and self-stretching during the non-bleeding period to prevent and reduce recurrent bleeding.
Treatment of bleeding should follow the PRICE principles of Prohibition, Rest, Ice, Compression, and Elevation.
(Elevation). In the case of muscle and joint bleeding, the PRICE principle is to infuse coagulation before the
The PRICE principle is an important management measure based on the infusion of clotting factors to raise clotting factor levels, and timely use of splints, molds, crutches, or The timely use of splints, molds, crutches or wheelchair braking can put the bleeding muscles and joints in a resting position, and the use of ice or cold wet compresses can effectively reduce the inflammatory response. It is recommended that ice be applied every 4 to 6 hours for about 5 to 10 minutes (no more than 10 minutes at a time) until swelling and pain are reduced.
In addition, a trained rehabilitation physician/therapist can assess the patient in terms of physical function, individual mobility, and social participation and, based on the results of the assessment, guide the patient through rehabilitation exercises to prevent, reduce, and minimize muscle and joint dysfunction and improve daily living. The patient’s assessment results are used to guide the patient’s rehabilitation to prevent, reduce, and minimize muscle dysfunction and improve daily activity and quality of life.
(a) Treatment of concurrent inhibitors.
The homo-neutralizing antibodies produced by HA patients receiving FⅧ replacement therapy are called suppressors, and the rate of suppressor production in heavy HA patients is 20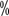 to 30
to 30 , and about 5 in medium or light HA
, and about 5 in medium or light HA to 10
to 10  chance of producing inhibitors. The continued presence of inhibitors is a serious complication of hemophilia, resulting in more difficult to control bleeding symptoms, increased risk of fatal bleeding, and a further reduction in quality of life. The presence of inhibitors is divided into two treatments: hemostasis and removal of inhibitors.
chance of producing inhibitors. The continued presence of inhibitors is a serious complication of hemophilia, resulting in more difficult to control bleeding symptoms, increased risk of fatal bleeding, and a further reduction in quality of life. The presence of inhibitors is divided into two treatments: hemostasis and removal of inhibitors.
- style=”margin-left: 59pt”>
- Hemostatic therapy.
Patients with acute bleeding should be treated with hemostasis as soon as possible. There are several ways to stop bleeding.
-
High dose FVIII: use only in patients with HA bleeding combined with low-titer inhibitors (≤5 BU/ml). The amount of FVIII required includes the amount used to neutralize the inhibitor and the amount needed to stop the bleeding. The amount of FVIII used to neutralize the inhibitor is calculated
“font-family:Times New Roman”>× 80 × [(1 – erythrocyte specific volume) × inhibitor titer (BU)]. To this, an additional 50 IU/Kg of FⅧ is required to ensure in vivo detection of FⅧ:C C is increased. If hemostasis is not effective, increase the dose or shorten the dosing interval, or switch to bypass therapy. In the case of low-titer, highly reactive inhibitors (inhibitor titer >5 BU/ml after another infusion of FVIII), consider switching to the bypass route after 3 to 5 days of dosing.- Bypass pathway agents: For combined high titer inhibitors (>5 BU/ml) or immune tolerance induction The bypass pathway agents are indicated for patients who have failed treatment with high titre inhibitors (>5 BU/ml) or immune tolerance induction (ITI) or who bleed during ITI treatment. Alternative “bypass pathway” drugs include activated prothrombin complex concentrate (aPCC) and recombinant activated coagulation factor VII (rF VIIa). rFⅦa The method of administration is 90μg/kg intravenously every 2-4 hours or 270. style=”font-family:Times New Roman”>μg/kg administered as a single dose. The recommended dose of PCC is 50-100 U/kg per dose at 8-12 hour intervals, not to exceed 200 U/kg per day in children with life-threatening bleeding, such as intracranial hemorrhage, who develop inhibitors. Once acute bleeding is stabilized, prophylactic therapy with PCC or rFⅦa is required for at least 6 months.
-
Emicizumab: Emicizumab prophylaxis It has been shown to be helpful in controlling bleeding, restoring target joint function, and improving quality of life in patients with hemophilia. The dosing regimen is the same as described above, with bypass agents discontinued 24 hours prior to dosing. If breakthrough bleeding occurs during prophylaxis, rFⅦa should be used as the first choice for treatment with an initial dose of ≤90. μg/kg for repeated dosing with a treatment interval of
should be greater than 2 hours. Also to avoid thrombosis, aPCC or PCC analogs should be avoided. FⅧ may also be used to treat breakthrough bleeding in patients with combined low-titer inhibitors.
- style=”margin-left: 59pt”>
- Clear suppressors.
ITI is the only recognized method of inhibitor clearance in inhibitor-positive patients who have been treated regularly and frequently with coagulation factor agents over a long period of time to achieve peripheral immune tolerance.HA combined with inhibitor-positive patients has an ITI success rate of approximately 70%.
-
Timing of ITI initiation: There is no international consensus on when the best time to start ITI is, but the current preference is to start ITI once diagnosed, regardless of inhibitor titer. ITI should be started as soon as the diagnosis is confirmed, regardless of the inhibitor titer.
span>VIIIconcentrated preparation or rhFVIIIare available, there is no evidence to suggest which formulation is superior, but when using rhFVIII style=”font-family:Arial”>for ITI therapy is unsuccessful, a switch to vWF-rich blood-derived FVIIIconcentrate may be considered. :Arial”>Concentrated preparations. - ITI regimen: i) First-line treatment:i High dose: 200 IU/(kg– d);
Increases in inhibitor titers or decreases in inhibitor titers of less than 20% over 6 months should be followed by gradual increases in ITI dose up to 200 IU/(kg–d); if the dose has reached 200 IU/(kg–d), a change to a second-line regimen is recommended. ②Second-line therapy: There is no standard second-line therapy. Consider switching to a different FⅧ product, such as switching from rhFⅧ to blood-derived FVIII, or in combination with human-derived CD20 monoclonal antibody clearance inhibitors, but further evaluation of long-term efficacy and safety is needed.
- style=”margin-left: 83pt”>
-
ITI efficacy assessment criteria: ① Fully tolerated. Persistent negative inhibitor
(<0.6 BU/ml) and FⅧ recovery >66%, FVIII half-life > 6 hours; (ii) Partially tolerated: inhibitor titer <5 BU/ml, although FVIII recovery is less than 66% and/or half-life < 6 hours, but treatment with FⅧ can stop bleeding; (3) Ineffective: full or partial tolerance cannot be achieved. In general, inhibitor titers fall by less than 20% within 3-6 months or remain >5 BU/ml after 3-5 years of ITI.
-
Prediction of ITI efficacy: It is currently believed that ITI efficacy is likely to be better in patients with the following characteristics: ① Inhibitor titer <10 BU/ml before starting ITI (ii) historical peak inhibitor titers <10 BU/ml before the start of ITI; (iii) peak inhibitor titers <100 BU/ml during ITI; (iv) time from diagnosis to the start of ITI <5 years; and (v) no interruptions since the start of ITI.
And ITI outcomes may be poor in patients with (i) inhibitor titers ≥10 BU/ml prior to initiation of ITI; (ii) historical peak inhibitor titers ≥200 BU/ml; (iii) peak inhibitor titers >100 BU/ml during ITI; (iv) time from diagnosis to initiation of ITI >5 years. (iv) >5 years from diagnosis to the start of ITI; (v) >2 weeks interval after the start of ITI.
- style=”margin-left: 72pt”>
-
Timing of termination of ITI: those who have reached full tolerance Move to prophylaxis
-
Treatment for those who have reached full tolerance; for those who have reached partial tolerance, if they can apply F span style=”font-family:Times New Roman”>VIII to adequately treat and stop bleeding symptoms, ITI therapy may be considered discontinued; failure to reduce inhibitor titers by 20
 or if ITI therapy has not been fully or partially tolerated for 3 to 5 years.
or if ITI therapy has not been fully or partially tolerated for 3 to 5 years.(ii) Management of hemophilic arthropathy.
Hemophilic arthropathy is a serious complication common in patients with hemophilia due to recurrent joint bleeding that leads to impaired joint function or joint deformity. To protect the joints and avoid disability, effective tertiary preventive therapy and multidisciplinary treatment need to be initiated immediately. Regular physical therapy and rehabilitation with regular joint structure [x-ray, MRI] is required while the patient maintains a certain FVIII valley concentration.
(MRI), ultrasound] and functional assessment. Analgesics may be applied appropriately to reduce pain, and orthopedic procedures such as synovectomy and arthroplasty may be performed depending on the condition. If surgery is to be performed, a comprehensive care team of experienced hematology specialists, orthopedic specialists, bleeding/coagulation laboratory technicians, and rehabilitation physicians is necessary to ensure perioperative evaluation of patient indicators, determination and smooth implementation of surgical protocols, and postoperative rehabilitation.
(iii) Management of hemophilic pseudotumors.
Hemophilic pseudotumor is a rare but fatal complication of hemophilia. It is essentially a cystic encapsulated hematoma that occurs in the muscle or bone, usually as a result of long-term chronic bleeding after inadequate clotting factor replacement therapy. The pseudotumor often encapsulates surrounding organs, making complete resection difficult. The perioperative and postoperative periods require multidisciplinary teamwork to prevent complications and pseudotumor recurrence.
(iv) Prevention and treatment of blood-borne diseases.
The common blood-borne viruses are human immunodeficiency virus, hepatitis C virus, and hepatitis B virus. Virus testing is recommended for hemophiliacs treated with blood-borne factor prophylaxis; hepatitis B vaccination is recommended for those who are HBsAb negative. In case of blood-borne viral infections, patients are recommended to undergo appropriate antiviral treatment under the guidance of the hemophilia multidisciplinary team. The use of genetic recombinants without any blood component eliminates the risk of infection from known and unknown pathogens.
Appendix 1.
 Table 1.
Table 1. Table 2.
Table 2. Table 3. Replacement therapy when access to coagulation factors is unrestricted
Table 3. Replacement therapy when access to coagulation factors is unrestricted< colgroup>
proof, can be extended)
Central nervous system/head
Start
80 to 100
1 to 7
Maintain
50
8 to 21
Throat and neck
Start
80 to 100
1 to 7
Maintain
50
8 to 14
Gastrointestinal
Start
80 to 100
7 to 14
Maintain
50
Kidneys
50
3 to 5
deep laceration
50
5 to 7
Surgery (large)
Pre-op
80 to 100
Post-op
60 to 80
1 to 3
40 to 60
4 to 6
30 to 50
7 to 14
Surgery (small)
Pre-op
50 to 80
Post-op
30 to 80
1 to 5 (depending on the type of surgery)
 Table 4. Replacement therapy when access to coagulation factors is limited
Table 4. Replacement therapy when access to coagulation factors is limited< colgroup>
Surgery (large)
Pre-op
60 to 80
Post-op
30 to 40
1 to 3
20 to 30
4 to 6
10 to 20
7 to 14
Surgery (small)
Pre-op
40 to 80
Post-op
20 to 50
1 to 5 (depending on the type of surgery)
Hemophilia A Guidelines (2022 Edition) Writing Validation Expert Group
Team leader:Huang Xiaojun
Members:Jing Wang, Haixia Fu, Lanping Xu, Qian Jiang, Hao Jiang, Xiaohui Zhang, Shenmiao Yang, Yuanyuan Zhang, Jin Song Jia, Xiaojun Huang, Jin Lu
- Bypass pathway agents: For combined high titer inhibitors (>5 BU/ml) or immune tolerance induction The bypass pathway agents are indicated for patients who have failed treatment with high titre inhibitors (>5 BU/ml) or immune tolerance induction (ITI) or who bleed during ITI treatment. Alternative “bypass pathway” drugs include activated prothrombin complex concentrate (aPCC) and recombinant activated coagulation factor VII (rF VIIa). rFⅦa The method of administration is 90μg/kg intravenously every 2-4 hours or 270. style=”font-family:Times New Roman”>μg/kg administered as a single dose. The recommended dose of PCC is 50-100 U/kg per dose at 8-12 hour intervals, not to exceed 200 U/kg per day in children with life-threatening bleeding, such as intracranial hemorrhage, who develop inhibitors. Once acute bleeding is stabilized, prophylactic therapy with PCC or rFⅦa is required for at least 6 months.