Chronic lymphocytic leukemia/small lymphocytic lymphoma guidelines
(2022 Edition)
I. Overview
Chronic lymphocytic leukemia/small lymphocytic lymphoma is a clonal proliferative neoplasm of mature B lymphocytes that presents with peripheral blood lymphocytosis, enlargement of the liver, spleen and lymph nodes, and involvement of organs other than the lymphatic system. In the late stage, it may manifest as bone marrow failure. Chronic lymphocytic leukemia (CLL) has the same pathological and immunophenotypic features as small lymphocytic lymphoma (SLL). The difference is that CLL disease is predominantly concentrated in the peripheral blood, whereas SLL disease is predominantly concentrated in the lymph nodes.
CLL/SLL is the most common type of leukemia in the West, accounting for 25 of all leukemias ~35
~35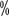 , with an annual incidence of (4 to 5)/100,000 in the European and American populations. The prevalence of CLL/SLL in Asian populations is significantly lower than that in Europe and the United States. The prevalence of CLL/SLL in Asian populations is significantly lower than in Europe and the United States. Population registries in Japan, Korea, and Taiwan show prevalence rates that are approximately 1/10th of those in Europe and the United States.
, with an annual incidence of (4 to 5)/100,000 in the European and American populations. The prevalence of CLL/SLL in Asian populations is significantly lower than that in Europe and the United States. The prevalence of CLL/SLL in Asian populations is significantly lower than in Europe and the United States. Population registries in Japan, Korea, and Taiwan show prevalence rates that are approximately 1/10th of those in Europe and the United States.
CLL/SLL tends to develop in older age, with the median age of onset reported in Europe and the United States being 70-75 years, whereas the median age of onset in China is 65 years.
II, Diagnosis and Differential Diagnosis
(A) Clinical manifestations.
style=”margin-left: 59pt”>
- Symptoms.
Early stages may be asymptomatic, and patients are often diagnosed by incidental blood count abnormalities on physical examination. Some patients may be seen for incidental painless enlargement of lymph nodes, mostly in the neck, which may sometimes recede and shrink on their own, but rarely disappear completely. In advanced stages, fatigue, night sweats, loss of appetite, hypothermia, and weight loss may occur. Acquired immunodeficiency may occur, and patients may have recurrent infections; or immune disorders such as autoimmune hemolytic anemia, immune thrombocytopenia, and pure red blood cell aplastic anemia may occur. Insect bite dermatitis is common.
style=”margin-left: 59pt”>
- Signs.
- Lymph node enlargement: There may be superficial lymph node enlargement, mostly in the neck and axilla, abdominal lymph node enlargement, and mediastinal lymph node enlargement. They may fuse and become large masses.
-
Spleen enlargement: often coexists with lymph node enlargement. A small number of patients with giant spleen may have left upper abdominal pain due to splenic infarction.
style=”margin-left: 72pt”>
- Hepatomegaly: may be present.
-
Wei’s ring swelling: narrowing of the oropharyngeal ring can be seen, either due to enlarged tonsils or It can be caused by lymphocyte infiltration in the submucosa causing thickening, which can trigger sleep apnea and dysphagia.
- Skin lesions: There can be leukemic skin infiltrates that require pathologic evaluation. There can be paraneoplastic syndrome manifestations such as aspergillosis and angioedema.
-
Other organ involvement: A small percentage of patients have nephrotic syndrome. Richter transformation, i.e., transformation to large cell lymphoma, can occur. Second tumors such as acute myeloid leukemia, myelodysplastic syndrome, skin cancer, lung cancer, gastrointestinal tract tumors, and melanoma can occur.
style=”margin-left: 59pt”>
- Laboratory tests.
-
Blood count: peripheral blood monoclonal B lymphocytes ≥5×109/L . The morphology of leukemic cells resembles that of mature small lymphocytes. Occasionally, primitive lymphocytes, a few naive or atypical lymphocytes are seen. The neutrophil ratio is reduced, and thrombocytopenia and/or anemia may develop as the disease progresses. The peripheral blood smear is easy to see.
The peripheral blood lymphocyte count is normal or mildly elevated in SLL, and the peripheral blood monoclonal B cells do not exceed 5×
. span>109/L.
Immune hematocrit and CLL progression-associated bone marrow failure need to be differentiated in hematocrit. Positive anti-human globulin tests may be seen. Biochemical and urine tests show signs of hemolysis.
Bone marrow and lymph node examination: bone marrow cytology with marked or extremely active nucleated cell proliferation and lymphocytes ≥40  , with predominantly mature lymphocytes; erythroid, granulocytic and megakaryocytic lineages are reduced; juvenile erythrocytes may be compensated for in hemolysis. The prognosis is worst for the diffuse type.
, with predominantly mature lymphocytes; erythroid, granulocytic and megakaryocytic lineages are reduced; juvenile erythrocytes may be compensated for in hemolysis. The prognosis is worst for the diffuse type.
Immunophenotype: The typical immunophenotype of CLL is kappa(κ) or lambda(λ) restricted, CD5+, CD23+, CD19+, CD20+; some patients have an atypical immunophenotype. Some patients with atypical immunophenotypes, especially mantle cell lymphoma (MCL) can be CD5+, and for better differentiation, CD43, CD79b, CD22, sIgM, CD81, CD200, CD10 or ROR1 can be added to assist in the diagnosis. The classic CLL phenotype is CD43+, CD79b weak+, CD81-, CD200+, CD10-
and ROR1+. Immunohistochemical staining for Cyclin D1, SOX11, LEF-1 and/or fluorescence in situ hybridization (FISH) for t(11;14) is required to identify with MCL.
-
Cytogenetics: application of interleukin (IL)2/CpG co-incubated G-band karyotype Analysis of only 40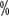 to 50
to 50 of CLL patients with chromosomal abnormalities. Complex karyotypic abnormalities can be detected. The detection rate can be increased to 80
of CLL patients with chromosomal abnormalities. Complex karyotypic abnormalities can be detected. The detection rate can be increased to 80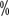 using FISH. Patients with initial diagnosis need to be tested for t(11;14), t(11q;v), and
using FISH. Patients with initial diagnosis need to be tested for t(11;14), t(11q;v), and
+12, 11q-, 13q-, 17p-, and other chromosomal abnormalities. Chromosomal abnormalities are important for the diagnosis, differential diagnosis, choice of treatment options, and prognosis of CLL. Patients with CLL with 13q- alone are the most common and have a better prognosis. The prognosis for patients with normal chromosomes and +12 is moderate, whereas patients with 11q- or 17p- have a poor prognosis, especially those with 17p-. New genetic abnormalities may arise during CLL disease progression, and patients with disease progression, relapse, or drug resistance need to be reevaluated cytogenetically.
-
Molecular biology:50  ~60
~60  Patients with somatic mutations in the immunoglobulin heavy chain variable region (IgHV) gene. CLL cells with IgHV mutations originate from memory B cells in the posterior germinal center, and such patients progress more slowly; CLL cells without IgHV mutations originate from primitive B cells in the anterior germinal center, and patients progress rapidly, are more likely to respond poorly to immunochemotherapy, and have a poor prognosis.
Patients with somatic mutations in the immunoglobulin heavy chain variable region (IgHV) gene. CLL cells with IgHV mutations originate from memory B cells in the posterior germinal center, and such patients progress more slowly; CLL cells without IgHV mutations originate from primitive B cells in the anterior germinal center, and patients progress rapidly, are more likely to respond poorly to immunochemotherapy, and have a poor prognosis.
IgHV fragment use also has prognostic significance. patients with IgHV3-21 application are suggestive of high risk regardless of IgHV gene mutation status.
TP53 mutations often occur in association with 17p-, but also in isolation. Because of the equally poor prognosis and susceptibility to treatment resistance, a new
Concomitant FISH and genetic testing is required before starting a new line of therapy. In addition, mutations in genes such as ATM, NOTCH1, BIRC3, and SF3B1 suggest a poor prognosis under immunotherapy treatment.
style=”margin-left: 59pt”>
- Imaging.
The clinical staging of CLL relies on palpation of the lymph nodes liver and spleen. Accurate measurements of lymph nodes and liver and spleen can be performed by B ultrasound, CT, etc. Accurate measurements of CT-based target lesions are required in outcome assessment. Positron emission tomography-computed tomography (PET-CT) is performed in patients with suspected Richter transformation and can provide assistance in biopsy pathology diagnosis.
(ii) Diagnostic criteria.
Polyclonal proliferation of cells. Heterotypic lymphocytes may be present.
-
Monoclonal B lymphocytosis: A healthy person with no symptoms or signs of peripheral blood B lymphocyte increased, but the count<5×109/L, without hepatosplenomegaly or lymph node enlargement.
-
Young gonocytic leukemia: increased number of young gonocytes, medium to large lymphocytes, and moderate amount of cytoplasm. The cytosolic volume is moderate, nucleoli are visible, and the count is ≥55 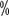 . sIgM is weakly expressed and CD5 is usually negative.
. sIgM is weakly expressed and CD5 is usually negative.
-
MCL: CD5, CD20 positive, CD20 and SIg strongly expressed, CD23 CyclinD1 or SOX11 helps to identify. t(11;14) positive by FISH.
-
Other B lymphocyte proliferative disease (B-LPD) The difference between B lymphocyte proliferative disease (B-LPD): see Table 1.
Table 1 Differential diagnosis of CLL and other B-LPD
|
Features
|
CLL
|
B-PLL
|
HCL
|
MCL
|
SMZL
|
FL
|
|
Morphology
|
|
Cell Size
|
small
|
Medium
|
Medium/Large
|
Medium
|
small
|
very small
|
|
Chromatin
|
in blocks
|
dense
|
sparse/cottony
|
speckled
|
dense
|
dense
|
|
Kernel
|
none/small
|
Significantly
|
none
|
none/small
|
none
|
none
|
|
Nuclear shape
|
Rules
|
Rules
|
Kidney-shaped
|
Cut marks
|
Rules
|
Nuclear fracture
|
|
Cytoplasm
|
Very little
|
Medium
|
Rich/Fuzzy
|
Medium
|
Less
|
Very little
|
|
Immunophenotype
|
|
Points
|
4 to 5
|
0 to 2
|
0
|
1 to 2
|
0 to 2
|
0 to 1
|
|
CD5
|
++
|
-/+
|
Negative
|
++
|
+
|
–
|
–
< col style="width:70px"/>
|
CD23
|
++
|
-/+
|
Negative
|
-/+
|
-/+
|
-/+
|
|
sIg
|
Weak expressions
|
Strong expression
|
Strong expression
|
Strong expression
|
Strong expression
|
Strong expression
|
|
FMC7
|
-/+
|
++
|
++
|
++
|
++
|
++
|
|
CD79b
|
Weak expression
|
Strong expression
|
Moderate expression
|
Strong expression
|
Strong expression
|
Strong expression
|
|
Immunohistochemistry
|
|
CCNDl
|
Negative
|
Negative
|
Weak expression
|
Positive
|
Negative
|
Negative
|
|
LEF-1
|
Positive
|
Negative
|
Negative
|
Negative
|
Negative
|
Negative
|
|
FISH
|
|
13q-
|
40  ~50% ~50%
|
Present
|
none
|
Present
|
Present
|
none
|
|
11q-
|
20 
|
Present
|
none
|
Present
|
Present
|
none
|
|
+12
|
15 
|
Rare
|
rare/none
|
Rare
|
none
|
rare
|
|
17p-
|
10 
|
50%
|
none
|
Present
|
Rare
|
none/rare
|
|
t(11;14)
|
none
|
none
|
none
|
Present
|
none
|
none
|
|
t(14;18)
|
none
|
none
|
none
|
none
|
none
|
Present
|
|
7q-/+3
|
none
|
none
|
none
|
none
|
Present
|
none
|
Note: -: negative or <10% of patients positive; -/+: 10%-25% of patients positive; +: 25%- 75% of patients positive. ++: >75% of patients positive; B-PLL: B juvenile lymphoblastic leukemia; HCL: hairy cell leukemia; MCL: mantle cell lymphoma; SMZL: splenic marginal zone lymphoma; FL: follicular lymphoma. All of the above immunophenotypes refer to flow cytometric results.
III.
(i) Clinical staging.
style=”margin-left: 59pt”>
- CLL: Binet staging and Rai staging are shown in Table 2.
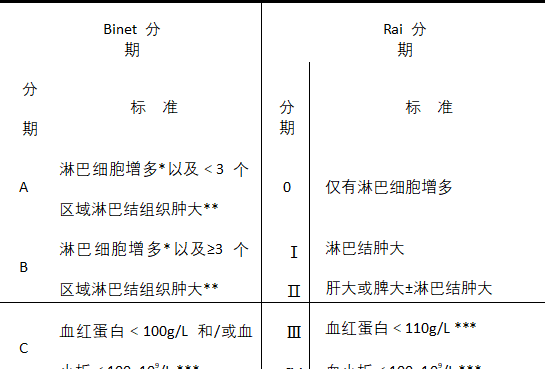
Table 2 Binet staging and Rai staging criteria for CLL
Note: *Absolute lymphocyte value ≥5×109/L.
**One region for each of the cervical, axillary, and inguinal lymph nodes on one or both sides, and one region for each of the liver and spleen, for a total of
Total 5.
** Excluding hemolysis and other causes of anemia or thrombocytopenia.
style=”margin-left: 67pt”>
- SLL: See Table 3 for Ann Arbor staging.

Table 3 Ann Arbor Staging Criteria for SLL

 Staging Lymph node involvement Extra nodal status early
Staging Lymph node involvement Extra nodal status early
I 1 lymph node or 1 group of lymph nodes Single extranodal lesion without lymph node invasion II Diaphragmatic side ≥ 2 groups of lymph nodes involved Stage I as determined by lymph node area
 or stage II then with extra-nodal invasion of adjacent lymph nodes
or stage II then with extra-nodal invasion of adjacent lymph nodes
Stage II large masses as above Stage II Standard plus “large masses” Not applicable


Late


III Bilateral lymph nodes of the diaphragm. Supradiaphragmatic lymph nodes with splenic involvement
Not applicable
 IV Stage III plus non-adjacent extra-nodal lymphoid tissue involvement
IV Stage III plus non-adjacent extra-nodal lymphoid tissue involvement
Not applicable
(ii) CLL International Prognostic Index.
The CLL international prognostic index (IPI) scores are shown in Table 4.

Table 4 CLL-IPI scoring system
IV.
(A) Indications for treatment.
-
Evidence of progressive bone marrow failure: demonstrated by a progressive decrease in hemoglobin and/or platelets with hemoglobin below 100g/L and platelets below 100
×109/L.
style=”margin-left: 48pt”>
- Massive spleen (e.g., >6 cm below the left costal margin) or progressive or symptomatic splenomegaly.
-
Massive lymph node enlargement (e.g., longest diameter >10 cm) or progressive or symptomatic lymph node enlargement.
style=”margin-left: 48pt”>
- Initial lymphocytes ≥30×109/L, with progressive lymphocytosis occurring.
If an increase of 50 , or a lymphocyte doubling time (LDT) <6 months. When the initial lymphocyte count is <30×109/L, LDT alone cannot be used as an indication for treatment. Other conditions that can cause lymphocytosis, such as infection or hormone application, need to be excluded.
, or a lymphocyte doubling time (LDT) <6 months. When the initial lymphocyte count is <30×109/L, LDT alone cannot be used as an indication for treatment. Other conditions that can cause lymphocytosis, such as infection or hormone application, need to be excluded.
- Autoimmune hemolytic anemia and/or thrombocytopenia does not respond well to corticosteroids or other standard treatments.
-
Extra-nodal lesions (e.g., skin, kidney, lung, spine, etc.) that are symptomatic or affect function, especially if symptomatic treatment does not resolve.
Severe fatigue (e.g., ECOG physical status ≥ 2; unable to perform routine activities).
style=”margin-left: 71pt”>
- No evidence of infection, temperature >38.0°C for more than 2 weeks.
- No evidence of infection and night sweats for more than 1 month.
Meet any 1 of the above to start treatment. Patients who do not meet the indications for treatment are followed up every 2 to 6 months with routine blood work, clinical symptoms, and liver, spleen, and lymph node enlargement.
(ii) Pre-treatment evaluation.
- The patient must be thoroughly evaluated before treatment: patients with CLL requiring treatment must be examined for the following items.
-
Present and past medical history: Include history of CLL treatment with details of comorbidities and current medications used for treatment.
style=”margin-left: 72pt”>
- Physical examination: especially of lymph nodes including pharyngeal lymphatic rings and liver and spleen
size.
style=”margin-left: 83pt”>
- Fitness status: as scored by ECOG.
(4) B symptoms: night sweats, fever, weight loss.
- Routine blood tests: including white blood cell count and classification, platelet count, hemoglobin, etc.
- Serum biochemical tests: including liver and kidney function, electrolytes, lactate dehydrogenase, β2 microglobulin, etc.
- Serum biochemical tests: including liver and kidney function, electrolytes, lactate dehydrogenase, β2 microglobulin, etc.
- Bone marrow biopsy±smear: pre-treatment, efficacy assessment and identification of the cause of cytopenias.
-
Hepatitis B surface antigen, hepatitis B surface antibody, hepatitis B e antigen, hepatitis B e antibody Hepatitis B core antibody, Hepatitis B virus DNA test.
-
CT of the neck, chest, abdomen, pelvis (recommended in cases of suspected large cell transformation) PET-CT, biopsy at the site of high metabolic uptake and refinement of pathology).
-
If Bruton’s tyrosine kinase is proposed ( Bruton‘s tyrosine kinase (BTK) inhibitor therapy, perform echocardiography and electrocardiography.
style=”margin-left: 79pt”>
- Pregnancy test in women of childbearing age.
- Karyotyping of peripheral blood, FISH for del(11q), and
+12, del(13q), del(17p) chromosomal abnormalities, and t(11;14) FISH can be performed to identify MCL. Complete peripheral blood TP53 mutation (NOTCH1, SF3B1, BIRC3, ATM, etc. when available), IgHV using fragment and mutation testing to determine prognosis and guide treatment.
-
Tests for special conditions: immunoglobulin quantification; reticulocyte count and direct anti-human globulin test; discussion of fertility and sperm bank related issues, etc.
(iii) First-line treatment options.
Because CLL/SLL remains an incurable disease, patients are first recommended to enter clinical trials in any case.
style=”margin-left: 48pt”>
- Treatment recommendations for patients without del (17p).
(1 ) <65 years of age and no serious concomitant disease [ cumulative disease score
(cumulative illness rating scale, CIRS) ≤ 6] patients: recommended: ① Ibrutinib, also consider Zebutinib, Orbutinib.
②fludarabine + cyclophosphamide + rituximab (for patients with mutations in the IgHV gene and younger than 65 years), bendamustine + rituximab (for patients with mutations in the IgHV gene and 65 years and older).
Other treatment options may be: vinblastine + otuzumab, fludarabine + rituximab, fludarabine + cyclophosphamide; or bendamustine monotherapy, fludarabine monotherapy, azacitidine pendentate ± rituximab.
-
Patients ≥65 years of age or <65 years of age with severe concomitant disease (CIRS >6).
Recommendations: 1) Ibrutinib, also consider Zebutinib, Orbutinib, Vinecla + Otuzumab; 2) Phenylbutyrate + Rituximab, Bendamustine + Rituximab.
Other treatment options may also be: rituximab, otuzumab, bendamustine monotherapy, fludarabine monotherapy, and pendimethalin monotherapy.
style=”margin-left: 71pt”>
- Debilitated patients (intolerant to purine analogs): ① Ibrutinib.
Zebutinib and obutinib can also be considered; ②Phenybutyric acid nitrogen mustard + rituximab.
Other treatment options could also be: vinecla + rituximab/otuzumab, rituximab monotherapy, otuzumab; or pembrolizumab monotherapy.
style=”margin-left: 59pt”>
- Patients with del (17p).
Recommendation: ibrutinib, also consider zebutinib, obutinib, enrolled in clinical trials.
Other treatment options are: high-dose methylprednisolone ± rituximab
/otuzumab.
(iv) Treatment options for relapsed, refractory patients.
Recommendations: (i) clinical trials; (ii) ibrutinib, zebutinib, obrutinib, vinblastine + rituximab; (iii) previous treatment remission ≥3 years can be considered to repeat the original regimen.
Other recommendations: any of the regimens mentioned in first-line therapy can be chosen based on the patient’s general status, such as fludarabine + cyclophosphamide + rituximab ± ibrutinib (for those with IgHV gene mutations and younger than 65 years of age), bendamustine
+Rituximab±Ibrutinib (for those with IgHV gene mutation and 65 years of age and older), high-dose methylprednisolone+Rituximab, otuzumab, lenalidomide±Rituximab, and participation in clinical trials.
(v) Hematopoietic stem cell transplantation.
Autologous hematopoietic stem cell transplantation is not recommended.
Allogeneic HSCT is a curative tool for CLL and can be considered in young patients without contraindications to transplantation who have the following indications
Cell transplantation: (i) in patients who have failed second-line or higher therapy including first-line BTK inhibitors or Bcl-2 inhibitors; and (ii) in patients with Richter syndrome.
(vi) Histologic transformation.
Patients with CLL who have transformed to diffuse large B cells, with consistent cellular origin identified by sequencing of the IgHV gene, have a poor prognosis, with a median survival of mostly less than 1 year, and treatment recommendations are based on the treatment regimen for aggressive lymphoma, with partial remission or better. Allogeneic hematopoietic stem cell transplantation is recommended for the treatment of aggressive lymphoma with partial remission or better. Diffuse large B-cell lymphoma of a different origin than CLL should be treated as diffuse large B-cell lymphoma. Patients with Hodgkin’s lymphoma transformation are treated as Hodgkin’s lymphoma.
(vii) Complication management.
-
Autoimmune hemocytopenia: hormonal therapy is recommended, with the option of intravenous gammaglobulin as first-line therapy. Refractory relapsed patients can be treated with rituximab, cyclosporine, and splenectomy.
-
Infection: Prevention and treatment of infection includes prevention and treatment of viral, bacterial, and fungal infections before and after CLL chemotherapy; and treatment of hepatitis B virus carriers. Anti-viral prophylaxis is required in the treatment of hepatitis B carriers, and human gammaglobulin infusion replacement therapy in patients with hypogammaglobulinemia. Once it occurs, aggressive pathogen screening, empiric anti-infective therapy, and subsequent selection of antimicrobial drugs based on positive culture and drug sensitivity test results.
-
Tumor lysis syndrome: For patients at high risk of developing tumor lysis syndrome, closely For patients at higher risk of tumor lysis syndrome, relevant blood parameters (potassium, uric acid, calcium, phosphorus and lactate dehydrogenase, etc.) should be monitored closely, along with adequate hydration and alkalinization, and the application of allopurinol or febuxostat to lower uric acid. In particular, patients treated with Vinecla should be graded for risk of tumor lysis syndrome and given appropriate preventive measures. Once it occurs, the
Adequate hydration and alkalinization with allopurinol, febuxostat, or labridase to lower uric acid, correction of electrolyte disturbances, and hemodialysis if necessary. Treatment.
(H) Efficacy criteria.
The efficacy criteria are shown in Table 5.
Complete response (CR): all criteria in Table 5 were met, with no disease-related symptoms.
Complete CR (CR with incomplete blood count recovery, Cri): meets CR criteria except that myelodysplasia has not returned to normal.
Partial response (PR): at least 2 group A criteria + 1 group B criteria.
PR with lymphocytosis (PRL): lymph node and spleen shrinkage meeting PR criteria after treatment with small molecule inhibitors of the B-cell receptor signaling pathway, accompanied by transient elevation of lymphocytes.
Stable disease (SD): No disease progression with failure to achieve PR.
progressive disease (PD): meeting any 1 of the Group A or B criteria.
Relapse: patient achieves CR or PR, ≥6 months after PD.
Refractory: treatment failure (no CR or PR) or <6 months PD after last chemotherapy.
Minimal residual disease (MRD) negative: <10-4 residual leukemia cells in peripheral blood or bone marrow.
Table 5 Efficacy criteria
|
Parameters
|
CR
|
PR >
|
PR-L
|
PD
|
|
Group A: for evaluation of tumor load
|
|
Lymph node enlargement
|
none>1.5 cm
|
Reduced by ≥50 
|
Reduced by ≥50 
|
Increase ≥50 
|
|
Large liver
|
none
|
Reduced by ≥50 
|
Reduced by ≥50 
|
Increase ≥50 
|
|
Spleen size
|
none
|
Reduced by ≥50 
|
Reduced by ≥50 
|
Increase ≥50 
|
|
Bone marrow
|
Normal hyperplasia, lymphocyte ratio
<30  , without B-cell lymph nodes; hypoplasia is CR with incomplete restoration of bone marrow hematopoiesis , without B-cell lymph nodes; hypoplasia is CR with incomplete restoration of bone marrow hematopoiesis
Recovery
|
Bone marrow infiltration decreased ≥50 percent from baseline  , or the presence of B-cell lymphoid nodules , or the presence of B-cell lymphoid nodules
|
Bone marrow infiltration decreased ≥50 percent from baseline  , or the presence of B-cell lymphoid nodules , or the presence of B-cell lymphoid nodules
|
|
|
peripheral blood lymphocytes
|
<4×109/L
|
Decrease from baseline by ≥
50 
|
Elevated lymphocytes
|
Increased from baseline by ≥
50 
|
|
Group B: Evaluation of bone marrow hematopoietic function
|
|
platelets (without growth factors)
|
>100×109/L
|
< span style="font-size:12pt">>100×< span style="font-family:Arial">109 /L or
Rise ≥ 50 from baseline 
|
>100×109/L or
Rise ≥ 50 from baseline 
|
Due to CLL native decline of ≥50 
|
|
Hemoglobin (no transfusion, no use
|
>110 g/L
|
>110 g/L or ≥
|
>110 g/L or ≥
from baseline |
Due to CLL the disease declined >20 g/L
|
|
Growth Factor)
|
|
50 
|
50 
|
|
|
peripheral blood neutrophils (without growth factors)
|
>1.5×10< span style="font-size:6pt">9/L
|
>1.5×109/L or
Higher than baseline>50 
|
>1.5×109/L or
Higher than baseline>50 
|
|
Appendix.
Chronic lymphocytic leukemia/small lymphocytic Lymphoma Treatment Guidelines (2022 Edition)
Writing and Validation Expert Group
(sorted by surname stroke)
Team leader:Huang Xiaojun
Members:Jing Wang, Haixia Fu, Lanping Xu, Qian Jiang, Hao Jiang, Xiaohui Zhang, Shenmiao Yang, Yuanyuan Zhang, Jin Song Jia, Xiaojun Huang, Jin Lu
 .
. of B-cell sIg is not expressed.
of B-cell sIg is not expressed. .
.